
Remember me
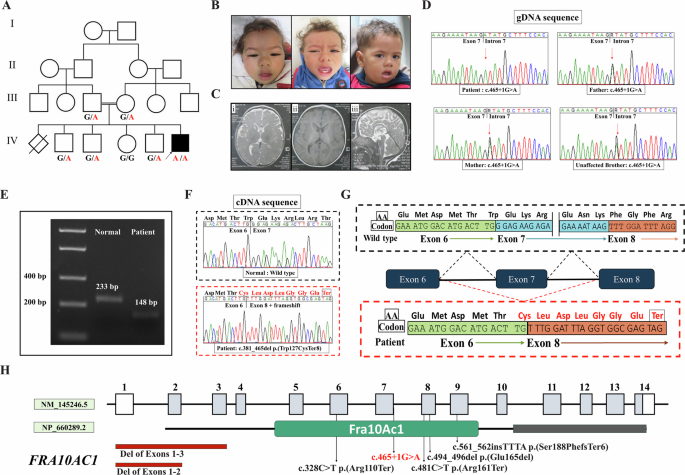
The 2-year-old girl was diagnosed with GDD and microcephaly at the age of 21 months. There was no family history of these conditions. The proband was delivered at full term through a normal and uncomplicated delivery process. The proband presented as a full-term small-for-gestational-age (SGA) infant who achieved the milestone of independent sitting at 14 months and independent walking at 21 months. At 18 months, the child demonstrated intentional vocalization of “Dad” and “Mom”. At 26 months, she possessed a vocabulary of 20–30 words but could not construct complete sentences. The proband underwent developmental evaluation using the Gesell Developmental Schedules at 19 months. The assessment revealed a global developmental age of 9.7 months, with an overall developmental quotient (DQ) of 49, indicative of moderate developmental delay. Domain-specific results were as follows: adaptive behavior (DQ = 59, developmental age at 11.6 months), gross motor skills (DQ = 37, developmental age at 7.3 months), fine motor skills (DQ = 52, developmental age at 10.3 months), language (DQ = 49, developmental age at 9.6 months), and personal-social skills (DQ = 50, developmental age at 9.8 months). These findings confirm profound delays at multiple aspects, particularly in gross motor and language function, consistent with her clinical presentation of global developmental delay.
Following physical examination, her anthropometric measurements were as follows: weight (W), 11 kg (3rd–10th percentiles); height (H), 90 cm (25th–50th percentiles); and head circumference (HC), 43 cm (<3rd percentile). She exhibited microcephaly, spasticity, multiple congenital facial malformations such as depressed nasal bridge, overhanging nasal tip, wide nasal base, large ear shape, and posteriorly rotated ears (depicted in Fig. 1A). In addition, she displayed slurred speech and abnormalities in dental morphology. She had never experienced seizures, aggressive behavior, self-mutilating behavior, autistic behaviors, attention deficit, or hyperactivity disorder. She underwent surgical intervention for coarctation of the aorta and patent ductus arteriosus.
Fig. 1
Facial appearance of the proband and Sanger sequencing results. A The proband exhibited microcephaly, depressed nasal bridge, overhanging nasal tip, wide nasal base, and abnormalities in dental morphology. B The Sanger sequencing results revealed a homozygous intron mutation in the proband and heterozygous intron mutation in the parents (c.224+5 G > A). The red arrow indicates the position of the mutation
Other tests including liver and kidney function tests, blood glucose tests, brain MRI, electroencephalogram (EEG), chromosome analysis, and CNV analysis, revealed no significant abnormalities.
WES, Sanger sequencing, and kinship analysisA homozygous mutation (c.224+5 G > A) located in the second intron of the METTL5 gene was identified by WES. Sanger sequencing of the METTL5 gene further validated the presence of the c.224+5 G > A homozygous variant in the proband, whereas both of her parents were carriers of the heterozygous mutation (Fig. 1B). Comprehensive single nucleotide variant (SNV) loci analysis of the proband’s genome could not detect any significant long homozygosity regions, thereby suggesting that consanguinity between the parents is unlikely. Meantime, haplotype reconstruction suggested that proband inherited different gene haplotype from patents (Table s1). These results provided compelling evidence against the presence of consanguinity between the proband’s parents According to the ACMG guidelines for assessing mutation pathogenicity, c.224+5 G > A was classified as a “Variant of Uncertain Significance” (VUS) based on PM2 + PM3_supporting+PP3 criteria. This mutation has been documented in NCBI database as RCV001794604.3 and RCV004783986.1. Regrettably, there is lack of detailed clinical data and pathogenic mechanism analysis.
Minigene assayTo ascertain the pathogenicity of the intron mutation, an in vitro splicing assay using a minigene vector was designed. This vector incorporated both the WT and mutant c.224+5 G > A sequences of the METTL5 gene, spanning exon 1 to exon 3. Further, RT-PCR yielded a 477-bp amplicon from cells transfected with the WT plasmid (Fig. 2A, Line2). In contrast, cells transfected with the mutant c.224+5 G > A sequence produced a 362-bp amplicon (Fig. 2A, Line3). Sanger sequencing then confirmed the exact splicing site. The results demonstrated a skipping of exon 2, resulting from a 115-bp deletion in the mutated sequence (Fig. 2B).
Fig. 2
Minigene vector constructs for the in vitro splicing assay. A Electrophoretogram of RT-PCR products: Lane 1: DNA ladder (Marker). Lane 2: Wild-type (WT) plasmid, yielding a 477-bp amplicon. Lane 3: Mutant-type (MT) plasmid, yielding a 362-bp amplicon. B Sanger sequencing of the RT-PCR products, confirming that the mutation c.224+5 G > A resulted in a 115-bp deletion causing exon 2 skipping
RT-PCR assay in vivoFollowing the validation of the intronic variant using the minigene assay, we performed RT-PCR on the blood samples from the family, confirming the splicing alteration of METTL5 in vivo. The proband with the c.224+5 G > A homozygous variant exhibited a 206 bp fragment (Fig. 3A, Line2), while both parents showed heterozygous variants with two bands (206 bp and 321 bp) (Fig. 3A, Line3 and Line4). The negative control samples displayed a 321 bp fragment (Fig. 3A, Line5).
Fig. 3
In vivo RT-PCR validation of METTL5 exon 2 deletion in the proband. A Nucleic acid electrophoresis results: Lane 1: DNA ladder (Marker). Lane 2: The proband exhibited a 206 bp fragment with the homozygous variant. Lane 3 and Lane 4: The parents exhibited heterozygous variants with two bands at 206 bp and 321 bp. Lane 5: The negative control exhibited a 321 bp fragment without variation. B Sanger sequencing chromatogram alignment, confirming that the mutation c.224+5 G > A resulted in a 115-bp deletion causing exon 2 skipping. C Schematic view of the splicing events in this mutation
Further sequencing analysis was conducted for the PCR fragments. For the homozygous proband, only the sequencing result corresponding to exon 2 deletion was observed. In heterozygous deletion carrier parents, overlapping peak patterns in the exon 2 region were detected due to the presence of one allele with exon 2 deletion and one normal allele. The similar phenomenon has been reported in a previous study [18]. Normal exon 2 sequencing peak patterns were observed in negative control samples. The schematic view of the splicing events of this mutation is presented in Fig. 3C.
METTL5 protein: bioinformatics analysisThe results of the two splice site prediction tools were as follows: NNSPLICE 0.9 software showed a score of 0.99 for the WT and 0.48 for the MT. Similarly, NetGene2 software also indicated a score of 0.93 for the WT and 0.55 for the MT. Variations in scores were observed between the WT and MT in these bioinformatics analyses, and these results indicated that the c.224+5 G > A mutation might lead to disruption in the splicing process, predicting the mutation to be “pathogenic”. Furthermore, Mutation Taster tool predicted the c.224+5 G > A mutation as “pathogenic”.
Literature reviewThe METTL5 gene mutations implicated in GDD or ID have been reported in five previous reports, including c.344_345delGA in a Pakistani family [10], c.571_572delAA in a Yemenite family [10], c.182 G > A in an Afghan family [19], c.362 A > G in another Afghan family [20], and c.223_224del in an Iranian family [21]. These mutations are associated with a wide range of symptoms, including moderate to severe ID, microcephaly, facial deformities, speech impairment, delayed walking, and behavioral abnormalities [10, 19,20,21]. Based on these studies, the most prominent feature is moderate to severe microcephaly related-GDD or ID. The second common clinical manifestation was distinctive facial features, such as wide nasal base, large ear shapes, and rotated posteriorly ears. Some patients may display the presence of autistic traits, aggressive behavior, seizures, or self-mutilating behaviors. Detailed information on clinical phenotypes associated with METTL5 gene mutations reported thus far is summarized in Table 1.
Table 1 Clinical manifestations & genotypes of previously reported METTL5 mutation cases
Comments (0)