
Remember me


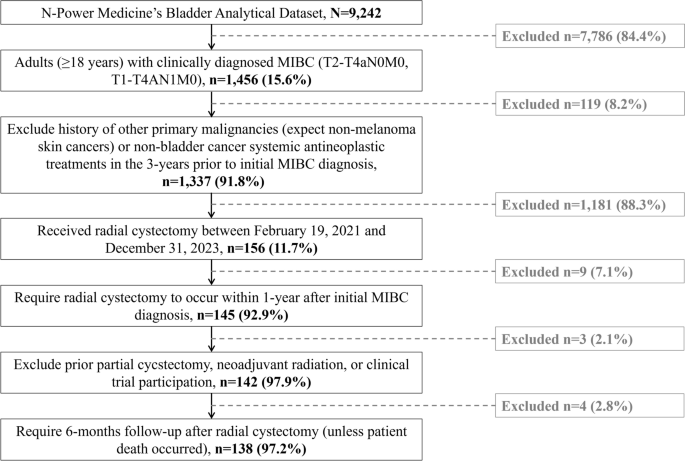






A total of 111 patients were retrospectively identified between 24 May 2022 and 30 January 2023 from 23 participating sites (first eligible patient identified on 5 July 2022). Among these, 108 (97.3%) met eligibility criteria and were included in the FAS, of whom 92 had HR+ disease and were included in the HRAS, and 74 initiated neratinib ≤ 1 year after completion of adjuvant trastuzumab-based therapy (median time 3.2 [range 0.3–11.9] months) and were included in the EMA-label subgroup (Fig. 1). The median follow-up time at data cut-off (5 July 2022) was 28.4 (range 4.4–57.9) months.
Fig. 1
Flow diagram of patient inclusion in the study. aThe HRAS included all patients in the FAS with HR+ disease (i.e. those with ER+ and/or PR+ disease at primary breast cancer diagnosis). bThe EMA-label subgroup included patients meeting the EMA-approved indication criteria (i.e. HR+ disease and initiation of neratinib ≤ 1 year since adjuvant trastuzumab-based therapy). EMA European Medicines Agency, ER+ oestrogen receptor-positive, FAS full analysis set, HER2+ human epidermal growth factor receptor 2-positive, HR+/− hormone receptor-positive/negative, HRAS hormone receptor-positive analysis set, PR+ progesterone receptor-positive
Patient Demographics and Clinical CharacteristicsIn the FAS, median age at neratinib initiation was 48 years, all patients but one were female, approximately one third of female patients were premenopausal and the majority had an Eastern Cooperative Oncology Group performance status of 0 (Table 1). Country of residence for patients in the FAS was Croatia (32.4%), Spain (22.2%), Italy (18.5%), Belgium (13.9%) or France (13.0%). Overall, 26 (24.1%) patients had one or more comorbidity at neratinib initiation, with hypertension being the most frequently reported (7/26 patients [26.9%]). Most patients in the FAS had stage II–III disease (74.1%), tumours with infiltrating ductal carcinoma histology (51.9%), ER+ and/or PR+ disease (84.3% and 68.5%, respectively) and lymph node involvement (41.7% with 1–3 nodes involved; Table 1).
Table 1 Patient demographics and clinical characteristics at neratinib initiationPatient demographics and clinical characteristics in the EMA-label subgroup were similar overall to those in the FAS (Table 1). The country of residence was Croatia (28.4%), Spain (28.4%), France (17.6%), Belgium (12.2%) or Italy (13.5%). As driven by the EMA label criteria, a higher proportion in the EMA-label subgroup than in the FAS had ER+ and/or PR+ disease (98.6% and 78.4%, respectively).
Most patients in the FAS (80.6%) and the EMA-label subgroup (81.0%) had a high risk of recurrence (i.e. stage II–III or node-positive disease, or non-pathological complete response after neoadjuvant therapy). Proportions of patients at low- versus high-risk were similar in the FAS and EMA-label subgroup (Supplementary Fig. S1).
Prior Breast Cancer Treatment HistoryRegarding previous therapies, more than half of patients in the FAS and EMA-label subgroup received both neoadjuvant and post-neoadjuvant treatment (58.3% and 64.9%, respectively; Table 1), with a median duration of neoadjuvant HER2-targeted therapy of 3.5 months in the FAS and 3.0 months in the EMA-label subgroup. In the neoadjuvant setting, approximately two thirds of patients in both the FAS and EMA-label subgroup received dual anti-HER2 therapy (64.9% and 68.2%, respectively; Fig. 2). Almost all patients who received neoadjuvant HER2-targeted therapy (FAS 98.2%; EMA-label subgroup 97.7%) completed this treatment as initially planned.
Fig. 2
Anti-HER2 neoadjuvant therapies received. Analyses included patients who received HER2-targeted treatment as neoadjuvant therapy. The EMA-label subgroup included patients with HR+ disease and initiation of neratinib ≤ 1 year from completion of trastuzumab-based adjuvant therapy. EMA European Medicines Agency, FAS full analysis set, HER2 human epidermal growth factor receptor 2, HR+ hormone receptor-positive
The most common type of breast surgery was mastectomy (in 60.2% in the FAS), followed by conservative surgery (lumpectomy [25.0%] or quadrantectomy [20.4%]). Axillary lymph node dissection was performed in 40.7% of patients and sentinel lymph node dissection in 25.0%. Post-surgery radiotherapy was received by almost all patients (90.7%) in the FAS.
Approximately 90% of patients in both the FAS and EMA-label subgroup reported having received HER2-targeted adjuvant therapy, either post-neoadjuvant or as adjuvant. In all, 10% of patients received chemotherapy ± endocrine therapy without trastuzumab. The most commonly used treatment was trastuzumab alone, administered to 87.3% and 88.4% of patients as post-neoadjuvant and adjuvant therapy, respectively, in the FAS group, and to 90.2% and 88.5%, respectively, in the EMA-label subgroup (Fig. 3).
Fig. 3
Anti-HER2 adjuvant therapies received in the a FAS and b EMA label subgroup. Analyses included patients who received HER2-targeted treatment as adjuvant therapy. The EMA-label subgroup included patients with HR+ disease and initiation of neratinib ≤ 1 year from completion of trastuzumab-based adjuvant therapy. Eight patients in the post-neoadjuvant group and two patients in the adjuvant group in the FAS did not receive anti-HER2 therapy; seven patients in the post-neoadjuvant group and zero patients in the adjuvant group in the EMA-label subgroup did not receive anti-HER2 therapy. EMA European Medicines Agency, FAS full analysis set, HER2 human epidermal growth factor receptor 2, HR+ hormone receptor-positive, T-DM1 trastuzumab emtansine
The median duration of post-neoadjuvant therapy in the FAS and EMA-label subgroup was 8.3 (range 0.0–21.0) and 7.7 (range 0.0–21.0) months, respectively, while that of adjuvant therapy was 11.8 (range 2.6–16.5) and 11.8 (range 9.0–13.0) months, respectively. T-DM1 as post-neoadjuvant therapy was received by only two patients each in the FAS and EMA label subgroup (Fig. 3).
The majority of patients (76.9%) in the FAS also received concomitant endocrine therapy, most frequently tamoxifen (44.6%) and letrozole (26.5%) (Supplementary Table S2).
Neratinib Treatment PatternsThe median time from diagnosis to initiation of neratinib therapy was 20.4 months in the FAS and 19.4 months in the EMA-label subgroup, with a median time from completion of adjuvant trastuzumab-based therapy to neratinib initiation of 4.6 and 3.2 months, respectively (Table 2). Most patients received an initial daily dose of 240 mg (87.0% and 86.5% in the FAS and EMA-label subgroup, respectively), with few patients in each subgroup receiving an initial dose of < 240 mg (13.0% in the FAS and 13.5% in the EMA-label subgroup). The median duration of neratinib treatment was approximately 12 months in both the FAS and EMA-label subgroup (12.3 and 12.1 months, respectively).
Table 2 Neratinib treatment patternsAs of 5 July 2022, 77 patients (71.3%) in the FAS and 50 patients (67.6%) in the EMA-label subgroup had completed neratinib treatment (Table 2). Overall, 67.6% of patients in the FAS and 64.9% in the EMA-label subgroup completed > 12 months of neratinib treatment, while 8.3% and 10.8%, respectively, had a treatment duration of ≤ 3 months.
The most common reason for premature treatment discontinuation was the occurrence of AEs (16.7% and 18.9% in the FAS and EMA-label subgroup, respectively). The frequency of premature treatment discontinuation was lower in patients treated with an initial daily neratinib dose of < 240 mg than in those with an initial daily dose of 240 mg (7.1% versus 25.5% in the FAS, respectively; Fig. 4 [10.0% versus 31.3%, respectively, in the EMA-label subgroup]). See Table 2 for more details on neratinib treatment patterns.
Fig. 4
Kaplan–Meier plot for time to discontinuation of neratinib, according to neratinib initial dose (FAS). Time to discontinuation was defined as the difference between the date of permanent discontinuation and the date of neratinib initiation + 1 day/30.4. For patients who were censored, the time was defined as the difference between the date of last available assessment and the date of neratinib initiation + 1 day/30.4. The longest neratinib duration observed was 16.4 months. The observation window was censored at 18 months. For three patients, the date of discontinuation was not available; these patients were censored at 1 day (i.e. 0.03 months). FAS full analysis set
SafetyRelevant AEsApproximately two thirds of patients in the FAS and EMA-label subgroup experienced at least one relevant AE (66.7% and 63.5%, respectively; Table 3). The safety profile of neratinib was similar in the FAS and EMA-label subgroup (Supplementary Table S3). Relevant AEs were mostly grade 1–2 in severity; grade 3 and 4 AEs were reported in 10.2% and 1.9% of patients, respectively, in the FAS, and 10.8% and 2.7% of patients, respectively, in the EMA-label subgroup (Table 3). In the FAS, SAEs were reported in 11.1% of patients (12.2% in the EMA-label subgroup), including one death of unknown cause. Most patients in the FAS and EMA-label subgroup fully recovered from relevant AEs (80.6% and 85.1%, respectively). A total of 93.1% of patients in the FAS (91.5% in the EMA-label subgroup) had relevant AEs that were attributed to neratinib.
Table 3 Relevant adverse eventsIn the FAS and EMA-label subgroup, the most reported AE of interest (any grade) was diarrhoea (58.3% and 52.7%, respectively) followed by other gastrointestinal disorders (29.6% and 24.3%; Table 3). Diarrhoea AEs were mostly grade 1 or grade 2 in severity (Supplementary Table S4). Grade 3 diarrhoea was reported in 6.5% of patients in the FAS, with grade 4 diarrhoea occurring in 1.9%. In the EMA-label subgroup, grade 3 and grade 4 diarrhoea were reported in 5.4% and 2.7% of patients, respectively. Among patients with diarrhoea, events were mostly considered to be related to neratinib (96.8% and 97.4% in the FAS and EMA-label subgroup, respectively) and led to temporary or permanent discontinuation of treatment in approximately one third of patients (27.0% and 30.8%, respectively; Supplementary Table S4). Diarrhoea AEs led to permanent treatment discontinuation in 11 (10.5%) and 8 (11.1%) patients in the FAS and EMA-label subgroup, respectively. Most patients with diarrhoea AEs recovered without sequelae (FAS 76.2%; EMA-label subgroup 79.5%).
The incidence and outcomes of diarrhoea AEs according to prior HER2-targeted therapy are summarized in Supplementary Table S4.
Prophylactic and/or Therapeutic Anti-diarrhoeal TreatmentApproximately half of the patients in the FAS (43.5%) and EMA-label subgroup (47.3%) received prophylactic anti-diarrhoeal treatment (Supplementary Table S5). Loperamide was by far the most commonly administered treatment, received by over 97% of patients. Diarrhoea of any grade occurred in a similar number of patients who received and did not receive anti-diarrhoeal prophylaxis; however, the severity of diarrhoea was lower with anti-diarrhoeal prophylaxis (grade 3 in two patients in the FAS and in one patient in the EMA-label subgroup, with no grade 4 in either subgroup) compared with those not receiving anti-diarrhoeal prophylaxis (grade 3 in five patients and grade 4 in two patients in the FAS; grade 3 in three patients and grade 4 in two patients in the EMA-label subgroup; Supplementary Table S5). Among those with diarrhoea AEs, the proportion of patients who discontinued neratinib (permanently or temporarily) due to diarrhoea was lower in patients who received prophylactic treatment than in those who did not.
Therapeutic anti-diarrhoeal treatment was received by approximately half of patients in the FAS (54.6%) and EMA-label subgroup (56.8%; Supplementary Table S6). As with its use in prophylaxis, loperamide was the most commonly reported treatment among patients who received therapeutic anti-diarrhoeal therapy. The proportion of patients who discontinued neratinib (permanently or temporarily) due to diarrhoea was lower in those who did versus did not receive therapeutic anti-diarrhoeal medication.
Exploratory EndpointsAfter a median follow-up of 26.2 months in the HRAS, seven patients (7.6%) experienced an iDFS event (5 [6.8%] after a median of 25.8 months in the EMA-label subgroup), with 24- and 36-month iDFS rates of 93.3% and 89.9%, respectively (94.5% and 89.5%, respectively, in the EMA-label subgroup; Table 4). Distant disease or death occurred in seven patients (7.6%) in the HRAS (5 [6.8%] in the EMA-label subgroup), with 24- and 36-month DDFS rates of 94.4% and 88.7%, respectively (95.8% and 87.9% in the EMA-label subgroup). No CNS metastases were observed in this study. The 24- and 36-month OS rates were 97.8% and 95.4% in the HRAS, and 97.2% and 94.1% in the EMA-label subgroup, respectively.
Table 4 Exploratory effectiveness analysis
Comments (0)