Hydrogel Fabrication and Fragmentation
Poly(ethylene glycol) diacrylate (PEGDA) (Sigma-Aldrich) and N-desulfated heparin methacrylamide (Hep−NMam) (Sigma-Aldrich) were synthesized as previously described [21]. Hydrogels containing poly(ethylene glycol) diacrylate (PEGDA), N-desulfated heparin methacrylamide (Hep−NMAm), and dithiothreitol (DTT; Sigma-Aldrich) were fabricated via free radical polymerization as previously described [29]. Briefly, PEGDA and DTT prepolymer solution was allowed incubate at 37 °C for 30 min. Prepolymer solution, including N-desulfated heparin, was crosslinked in a 1 mL syringe using thermal initiators ammonium persulfate (APS; Sigma-Aldrich, 248,614) and tetramethylethylenediamine (TEMED; Sigma-Aldrich, 411,019) with incubation at 37 °C for 30 min. Pre-polymer solution for fluorescently tagged fragments contained 0.33 mg/mL FITC-PEG-SH (1 kDa; NANOCS) as previously described [30]. After incubation, the hydrogels were passed through a 40 μm filter (PluriSelect) with DI water 4 times to make hydrogel fragments. Fragments were then lyophilized (Labconco) and kept at −20 °C until use.
In VitroSDF-1α Loading and Release
Hydrogel fragments with varying amounts of Hep−N were loaded in vitro by adding 1.30 μg human SDF-1α (R&D Systems) to 0.8 mg fragments in 400μL 0.1 wt% bovine serum albumin (BSA; Thermo Fisher) solution, and incubating the fragments and protein for 2 h at 4 °C. After incubation, the fragments were centrifuged (Eppendorf 5420), and the supernatant was removed to determine the amount of SDF-1α that was loaded onto fragments via human SDF-1α ELISA (R&D Systems; n = 4). To determine cumulative release over time, the supernatant was collected and replaced 3 h and 1, 3, 5 and 7 days following SDF-1α loading. The amount of SDF-1α in the collected supernatant was quantified via human SDF-1α ELISA (R&D Systems; n = 4).
Tendon Transection and Denervation
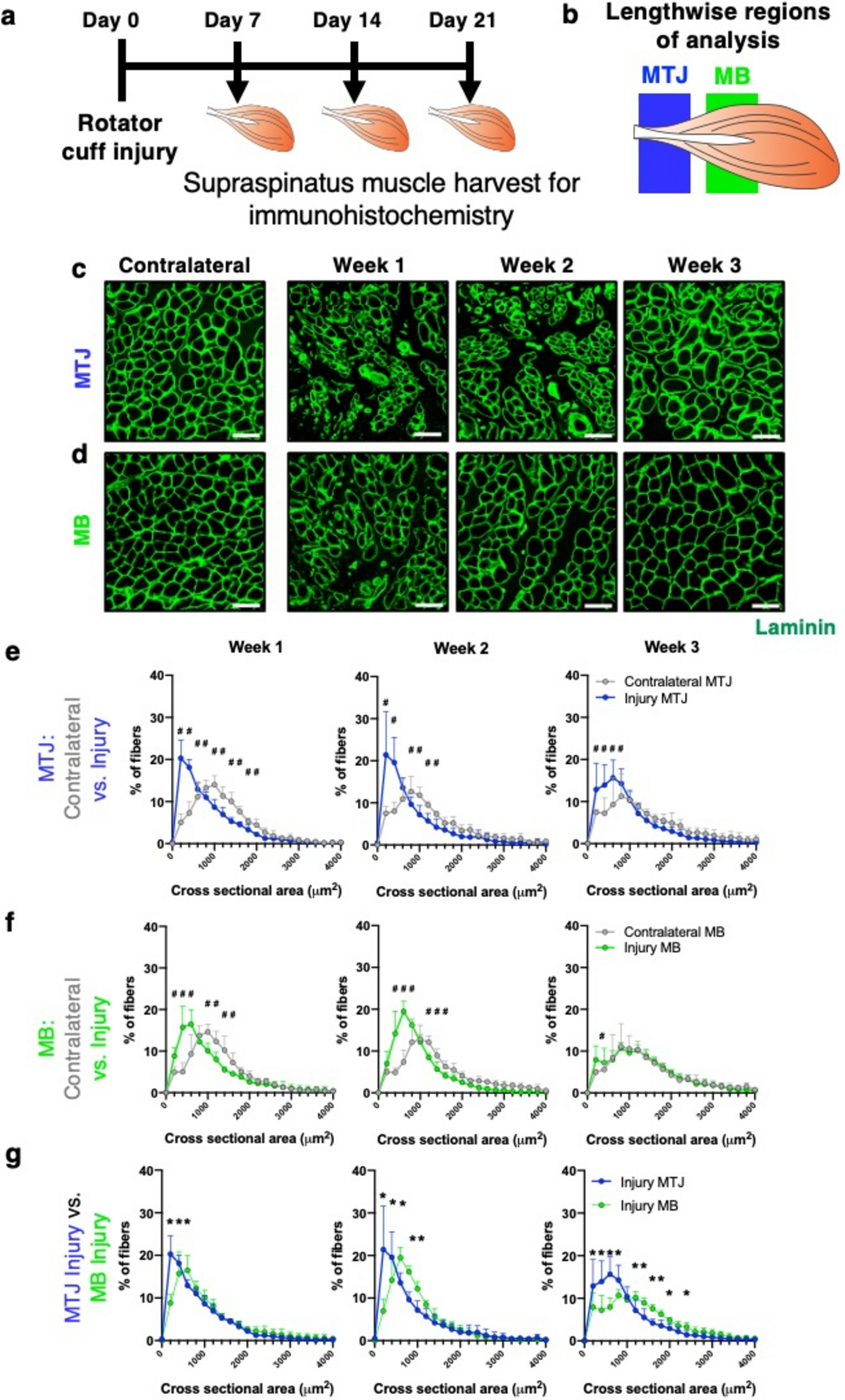
Rotator cuff injury was induced as previously described [20, 31, 32]. 8- to 10-week-old male Sprague–Dawley rats, initially weighing 250–300 g, were used in accordance with protocols approved by the Georgia Institute of Technology Institutional Animal Care and Use Committee. First, animals were initially anesthetized using 5% isoflurane (Isothesia). Anesthesia was maintained with 2–3% isoflurane during surgery. The left chest and forelimb were shaved and sanitized with alcohol and chlorhexidine. Next, a ~ 2 cm incision was made through the skin and deltoid muscle to expose the suprascapular nerve. To induce denervation, a ~ 0.5 mm segment of the suprascapular nerve was resected. Next, the supraspinatus and infraspinatus tendons were transected, surrounded with a 0.3 mm portion of PharMed BPT tubing (Tygon, 1.6 mm inner diameter) and secured with 4–0 silk non-absorbable sutures (Oasis), in order to prevent spontaneous reattachment of the tendons to the humeral head [33]. The deltoid was then closed using Vicryl 4–0 resorbable sutures (Ethicon) and the skin was closed using wound clips. Finally, immediately after surgery, sustained release buprenorphine was administered as an analgesic.
Fragment Loading and Injection Strategy
5% w/v hydrogel fragments containing 20 wt% Hep−N and 70 mol% DTT were utilized for all in vivo studies. To load SDF-1α onto fragments for in vivo cellular recruitment and histological studies, fragments to complete each surgery were resuspended with 0.1 wt% sterile BSA-Phosphate Buffered Saline (PBS; Corning, 70,011,044) solution containing sterile human SDF-1α, to a concentration of 80 mg/mL. The loading ratio of SDF-1α to fragments was 1.3 μg of SDF-1α to 0.8 mg fragments. Loaded fragments were allowed to incubate overnight at 4 °C and transferred to sterile Hamilton syringes with 22-gauge 1.5-in. hypodermic needles (BD, 305,156).
Immediately after injury and before closing the deltoid, 10uL (~ 0.8 mg) of SDF-1α loaded fragments, were injected 5 mm or 10 mm deep into the muscle, corresponding to the MTJ or MB regions of the muscle, respectively. Tygon tubing was used as a needle guide to ensure proper depth, and a needle track was created in each region of the muscle, regardless of injection location. Saline treatment was included as a control, where 20µL saline was injected throughout the MTJ and MB regions.
In vivoSDF-1α Release
For in vivo imaging of SDF-1α loaded fragments in each injection region, 6.4 µg AlexaFluor647-labelled SDF-1α (AF647 SDF-1α; R&D Systems) was loaded onto 2 mg fragments and allowed to incubate at 4 °C overnight. (Based on previous work [20], increased dosage was used for this study to ensure that the fluorescent signal was sufficient for detection by the instrument.) Fragments were then loaded into sterile Hamilton syringes. Immediately after injury, 20uL (~ 2 mg) of SDF-1α loaded fragments were injected into either the MTJ or MB region of the supraspinatus muscle. The left forelimb and back of the animals were shaved, and skin was sanitized using iodine and alcohol. Animals were imaged with IVIS on day 0, 1, 3, 7, 10, 14 and 21 following injury and injection. Total radiant efficiency [p/s]/[µW/cm2] was determined using the same-sized circular region of interest (ROI) at each timepoint and animal. Background fluorescence from the skin was evaluated for each animal at the final timepoint (day 21). An average of the day 21 background signal from all animals was subtracted from each ROI measurement. Fluorescent signal at each time point was normalized to the signal at day 1; n = 4–6.
Flow Cytometry
Supraspinatus muscles (n = 7–9 for each treatment group) were harvested and analyzed for inflammatory cell and stem cell populations using flow cytometry at days 3 and 7 (Fig. 1). 3 and 7 days were chosen as timepoints in this study because effects of SDF-1α treatment on cellular milieu have been observed in our model at these timepoints [20]. Day 3 and Day 7 saline control data sets were collected and reported previously [27].
After dissection, each muscle was split into MTJ (first 7 mm of muscle, in relation to the insertion region) and MB (remainder of muscle) regions. A digestion solution containing Dispase II (ThermoFisher, 17,105–041), Collagenase Type II (Worthington, LS004176) and Dulbecco’s modified Eagle medium (DMEM; ThermoFisher, 11,965,118) was used to digest dissected muscle portions for 1 h and 20 min at 37 ºC, and the resulting cell suspension was pipetted through a 40 μm cell strainer (Corning).
Each sample was split in half and stained with either an inflammatory cell or stem cell panel. The inflammatory cell panel included PE conjugated CD163 (Bio-rad, MCA342PE), PerCP-Cy5.5 conjugated CD3 (Biolegend, 201,418), PE-Cy7 conjugated CD4 (BD, BDB561578), APC conjugated CD86 (Biolegend, 200,315), AF700 conjugated CD68 (Bio-rad, MCA341AF700), Pacific Blue conjugated CD11b (Bio-rad, MCA275PB), BV605 conjugated RP-1 (BD, BDB743055), BV786 conjugated CD25 (BD, BDB742757), BV 480 conjugated CD8 (BD, BDB746832), and FITC conjugated CD206 (Bioss, 10,398–504).
Antibody preparation for the stem cell panel included conjugating SCA-1 (Fisher-Sigma, AB4336MI) with APC via APC Conjugation Kit – Lightning-Link® (Abcam, ab201807). The stem cell panel included FITC-conjugated PDGFRa (Bioss, BS-0231R-FITC), PE-conjugated CD106 (BD, BDB559229), PE-Cy7 conjugated CD29 (Biolegend, 102,222), APC-Cy7 conjugated CD45 (BD, BDB561586), BV421 conjugated CD90 (BD, BDB563770), BV605 conjugated CD31 (BD, BDB744539), BV68 conjugated CD44 (BD, BDB743925) and APC conjugated SCA-1 (see details above).
Samples were stained for 30 min and fixed in 2% paraformaldehyde (PFA, J.T. Baker) for 20 min, then analyzed using a Cytek Aurora spectral flow cytometer (Cytek Biosciences). The following cell types were identified: Leukocytes (CD11b +), neutrophils (CD11b + CD45 + RP-1 +), M1-like macrophages (CD11b + CD68 + CD86 +), M2c-like macrophages (CD11b + CD68 + CD163 +), M2a-like macrophages (CD11b + CD68 + CD206 +), T lymphocytes (CD3 +), cytotoxic T cells (CD3 + CD8 +), helper T cells (CD3 + CD4 +), regulatory T cells (CD3 + CD4 + CD25 +), mesenchymal stem cells (CD45-CD31-CD90 + CD29 + CD44 +), satellite cells (CD45-CD31-Sca1-VCAM-1 +), and fibro-adipogenic progenitor cells (CD45-CD31-Sca1 + VCAM-1-). For data analysis, each cell population was first calculated as a percentage of single cells by dividing the number of cells in each population by the number of single cells.
Spanning-tree Progression Analysis of Density-normalized Events (SPADE)
SPADE, a MATLAB based program, generates trees via four computational modules: density dependent downsampling, agglomerative clustering, connection of clusters via a minimum spanning tree algorithm, and cell mapping via upsampling [34]. Each tree consists of nodes that represent clusters of cells with similar marker expression. The size and color characteristics of each node are determined by the number of cells present as well as median surface marker expression.
SPADE was used to create population level trees for the expression of macrophage subtype markers CD86 (M1-like macrophages), CD206 (M2a-like macrophages), and CD163 (M2c-like macrophages) in CD11b + CD68 + cells. The SPADE trees were generated by exporting compensated and manually gated macrophage subsets (CD86 gating tree). MATLAB was used to run SPADE analysis, with the source code that can be found at the following website: http://pengqiu.gatech.edu/software/SPADE/. These SPADE parameters were applied: Arcsinh transformation with cofactor 5, neighborhood size 5, local density approximation factor 1.5, max allowable cells in pooled down sampled data 50,000, target density 20,000 cells remaining, and 20 desired clusters. Cell frequency overlays of each group were then compared via the compare groups function, with a p-value < 0.05.
Muscle sectioning for Immunohistochemical Stains
7 days after injury, injured supraspinatus muscles (n = 6 per treatment region) were harvested and prepared for histological analysis. Muscles were incubated in 10% v/v optimum cutting temperature (OCT; Sakura Finetek) PBS solution (PBS:OCT solution), PBS:OCT solution containing 10% sucrose (VWR), and PBS:OCT solution containing 20% sucrose for 30 min each to completely infiltrate the tissue. Then, muscles were embedded in 100% OCT and placed under vacuum overnight. A dry ice + ethanol was used to cool hexanes (Sigma-Aldrich) in a stainless-steel beaker and allowed to equilibrate for 30 min. Each block was then frozen in ice bath cooled hexanes. Muscles were sectioned into 10 μm cross-sections at 20–50 μm intervals with a cryostat (Thermo Scientific CryoStar NX70).
Immunohistochemical Staining of Muscle and Analysis
Muscle sections were immunostained for laminin and embryonic myosin heavy chain or perilipin and collagen and imaged as previously described [31]. Blocking buffer (2% bovine serum albumin (Thermo Scientific), 0.5% goat serum (R&D Systems), 0.5% Triton X (Amresco) in PBS solution) was applied to the sections for 30 min at room temperature. A wash step was performed between each step, during which 0.1 wt% Tween20 (VWR) in PBS solution was applied to slides three times for two minutes each. Primary antibodies for embryonic myosin heavy chain (eMHC) (Developmental Studies Hybridoma Bank, mouse monoclonal anti-eMHC, product code: F1.652-s) and laminin (Sigma, rabbit polyclonal anti-laminin, product code: L9393) were diluted 1:100 and 1:200 in blocking buffer, respectively. Primary antibodies for perilipin (Cell Signaling Technologies, rabbit monoclonal anti-perilipin-1, product code: 9349S) and collagen (Sigma, mouse monoclonal anti-collagen type-1, product code: SAB4200678)) were diluted 1:100 and 1:200 in blocking buffer, respectively. Primary antibodies were added to sections for one hour at room temperature and subsequently washed with PBS-Tween20 solution.
Secondary antibodies for eMHC (Goat anti-mouse IgG H + L (AlexaFluor568), Thermo Fisher, product code: A11004) and laminin (Goat anti-rabbit IgG H + L (AlexaFluor 488), Abcam, product code: ab150077) were diluted 1:250 in blocking buffer, added to sections for 30 min at room temperature, and washed with PBS-Tween20 solution. Secondary antibodies for perilipin (Goat anti-rabbit IgG H + L (AlexaFluor 488), Abcam, product code: ab150077) and collagen (Goat anti-mouse IgG H + L (AlexaFluor568), Thermo Fisher, product code: A11004) were diluted 1:250 in blocking buffer, added to sections for 30 min at room temperature, and washed with PBS-Tween20 solution.
Hoechst 33,342 fluorescent stain (Thermo Fisher, product code: 62,249) was diluted 1:50,000 in PBS-Tween20 solution, added to slides for 2 min, and washed with PBS-Tween20 solution. Sections were then mounted with Vectashield (Vector Laboratories) for imaging using a Zeiss LSM 710 confocal microscope with a 10 × objective.
All tissue level analysis was performed using ImageJ. eMHC + fiber quantification was performed on 1 section per region of each muscle, for 5 animals in each group. To determine the number of regenerating muscle fibers, the number of eMHC + fibers (defined as + 2 s of the mean pixel intensity per muscle fiber) was divided by total area of the muscle within 500 µm of the intramuscular tendon (near intramuscular tendon region).
Quantification of the density of centrally located nuclei (CLN) was performed on 1 section per region of each muscle, for 5 animals in each group. A centrally located nuclei was defined as having < 50% offset from the center in either the major or minor axis direction. CLN were only characterized in the near intramuscular tendon region. The muscle fiber cross-sectional area (CSA) quantification was performed on 1 section per region of each muscle, for 5 animals in each group. The CSA was characterized only for muscle fibers (segmented based on the laminin stain) in the near intramuscular tendon region.
Quantification of the percent of the entire muscle section positively stained for collagen was performed on 1 section per region of each muscle, for 5 animals in each group. Quantification of the percent of the entire muscle section positively stained for perilipin was performed on 1 section per region of each muscle, for 5 animals in each group. Perilipin and collagen staining associated with the muscle periphery and blood vessels was excluded from the analysis.
Statistics
All data are presented as mean ± standard deviation. Except SPADE, all data analysis was performed using Prism 10 software. Outliers were removed prior to statistical tests for IVIS and cellular recruitment studies using the ROUT method. Statistical analysis for in vitro SDF-1α loading consisted of one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test. Analysis for in vitro release of SDF-1α included two-way ANOVA with Tukey’s multiple comparisons test. Statistical analysis for flow cytometry data, in vivo SDF-1α release, and eMHC analysis consisted of two-way ANOVA followed by Tukey’s or Sidak’s multiple comparison test where appropriate. Significance value was set at p ≤ 0.05 for all tests.



Comments (0)