
Remember me
Bi-allelic SFTPB variants leading to surfactant protein (SP)-B deficiency are very rare and represent one of the most severe causes of childhood interstitial lung diseases (chILD).1 To date, more than 40 SFTPB pathogenic variants have been described, including the loss-of-function variation, c.361delinsGAA p.(Pro121Glufs*95), previously named 121ins2, identified in two-thirds of cases.2 Typical presentation is immediate and fatal respiratory distress syndrome (RDS), with or without pulmonary hypertension (PHT) at birth.3 Very few comprehensive SFTPB series have been reported so far. This study aimed to establish phenotype–genotype correlations of patients with SP-B deficiency in the French cohort and to guide management and genetic counselling.
MethodsPatients under 18 years diagnosed with SFTPB bi-allelic pathogenic or likely pathogenic variants were retrospectively identified in the French rare lung disease network RespiRare centres between 1995 and 2022.2
The data were collected from the patient records and included clinical and therapeutic information, initial thoracic imaging, echocardiography, histopathology and genetic analyses.
Qualitative/categorical variables were presented as numbers and percentages. Quantitative/continuous variables were presented as median and IQR. χ2 and Fisher’s exact tests were used to compare nominal variables, and Kruskal-Wallis or Mann-Whitney tests were used to compare ordinal variables. A p value≤0.05 was considered significant.
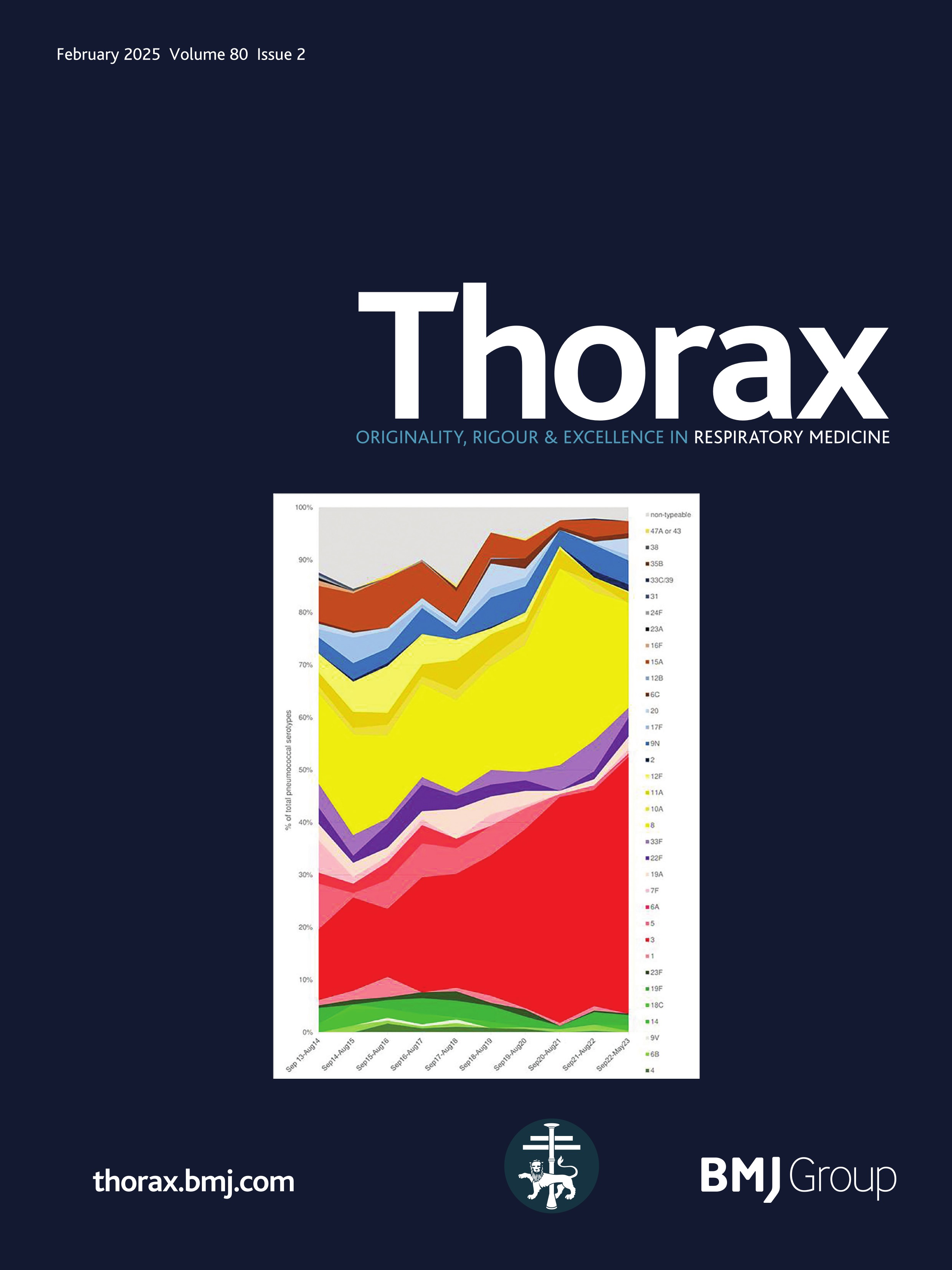
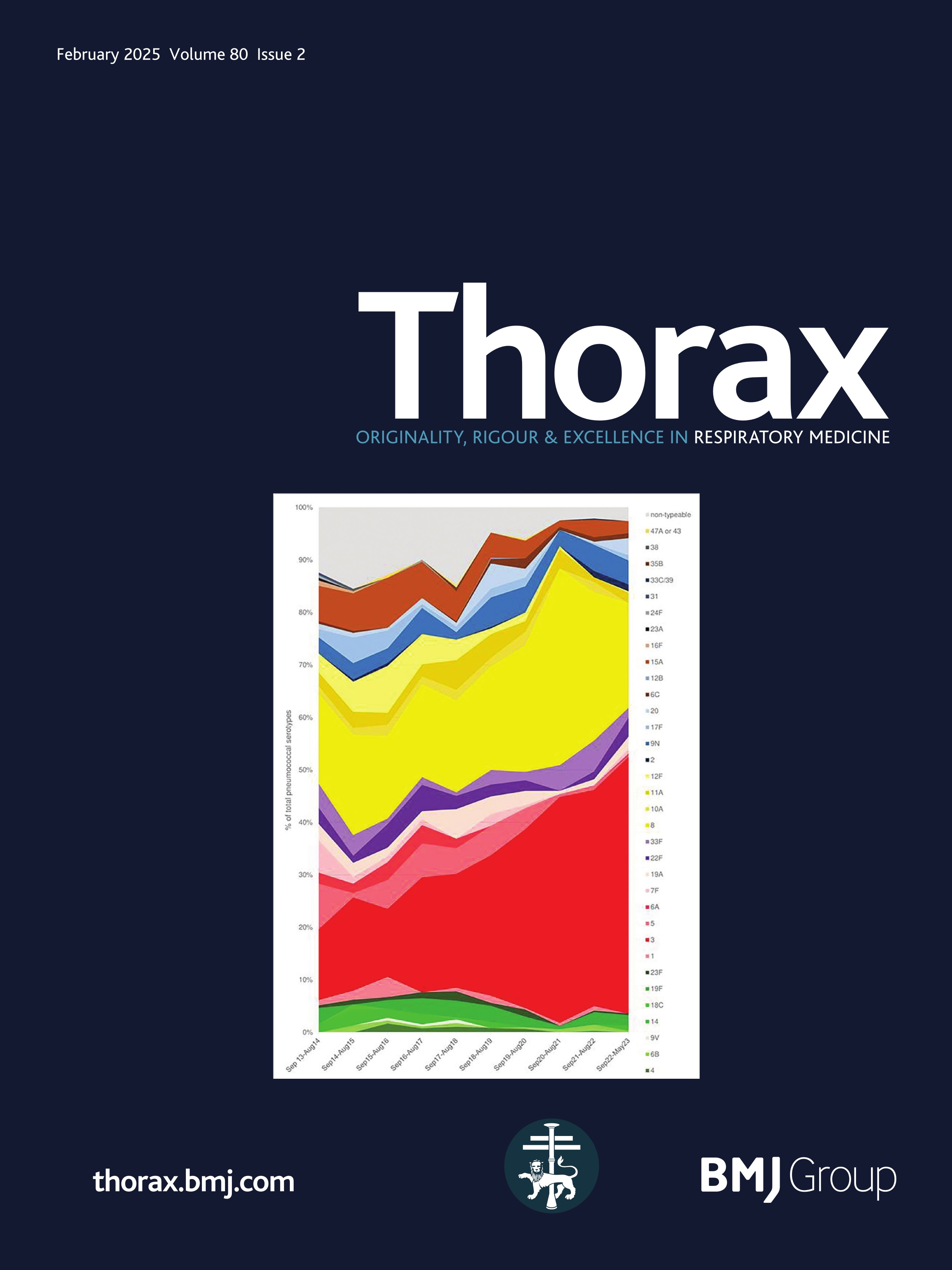
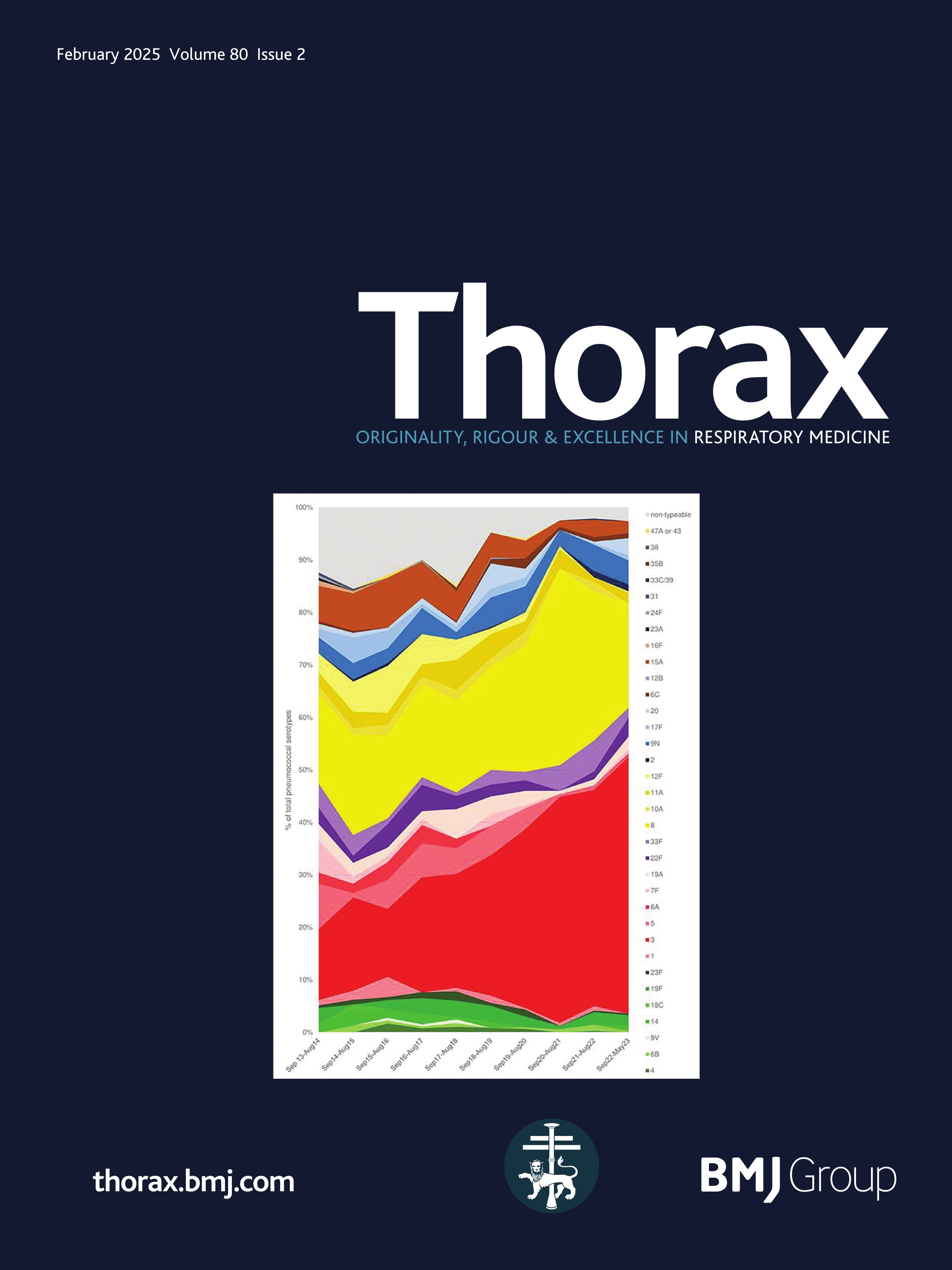
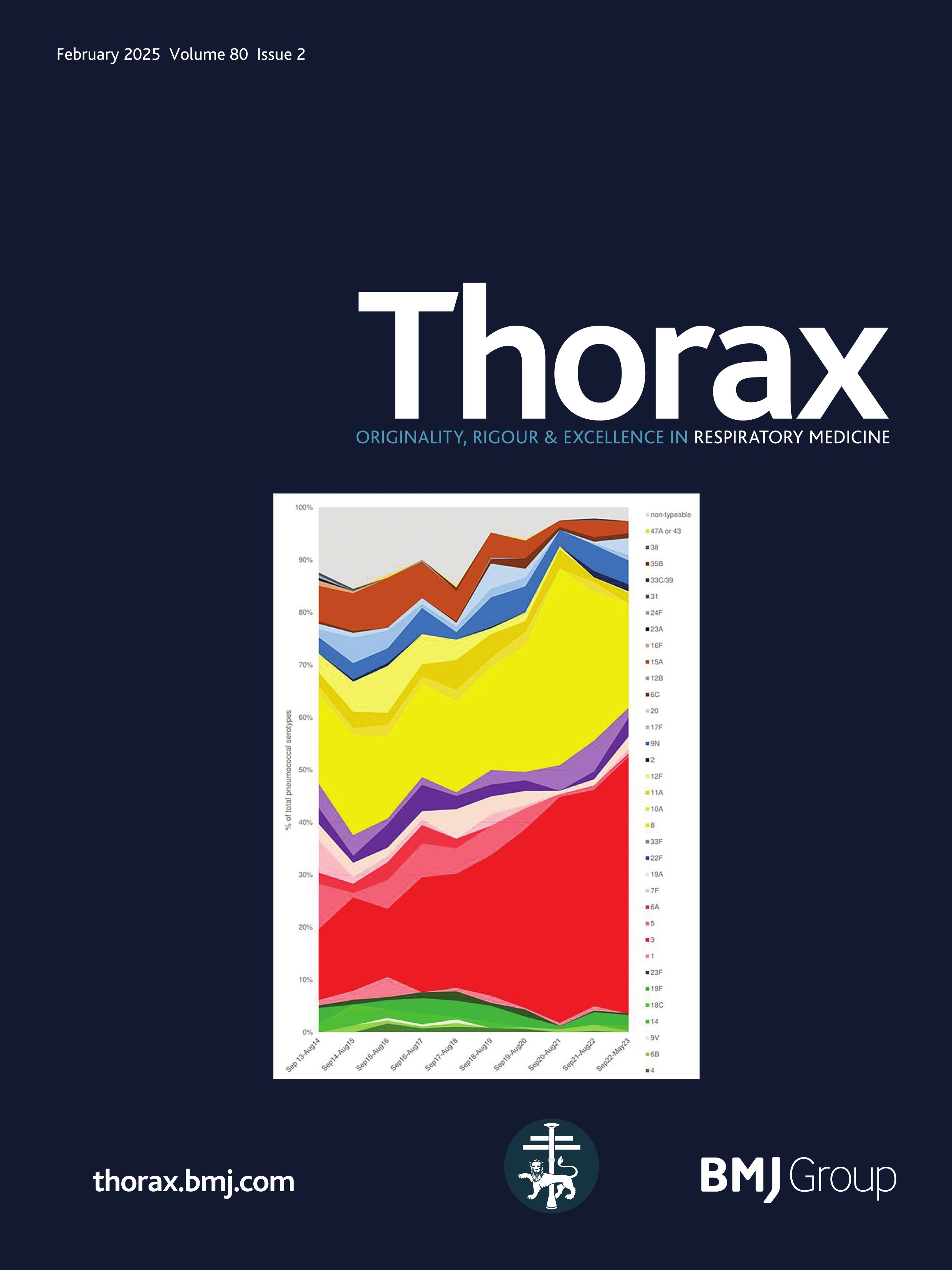
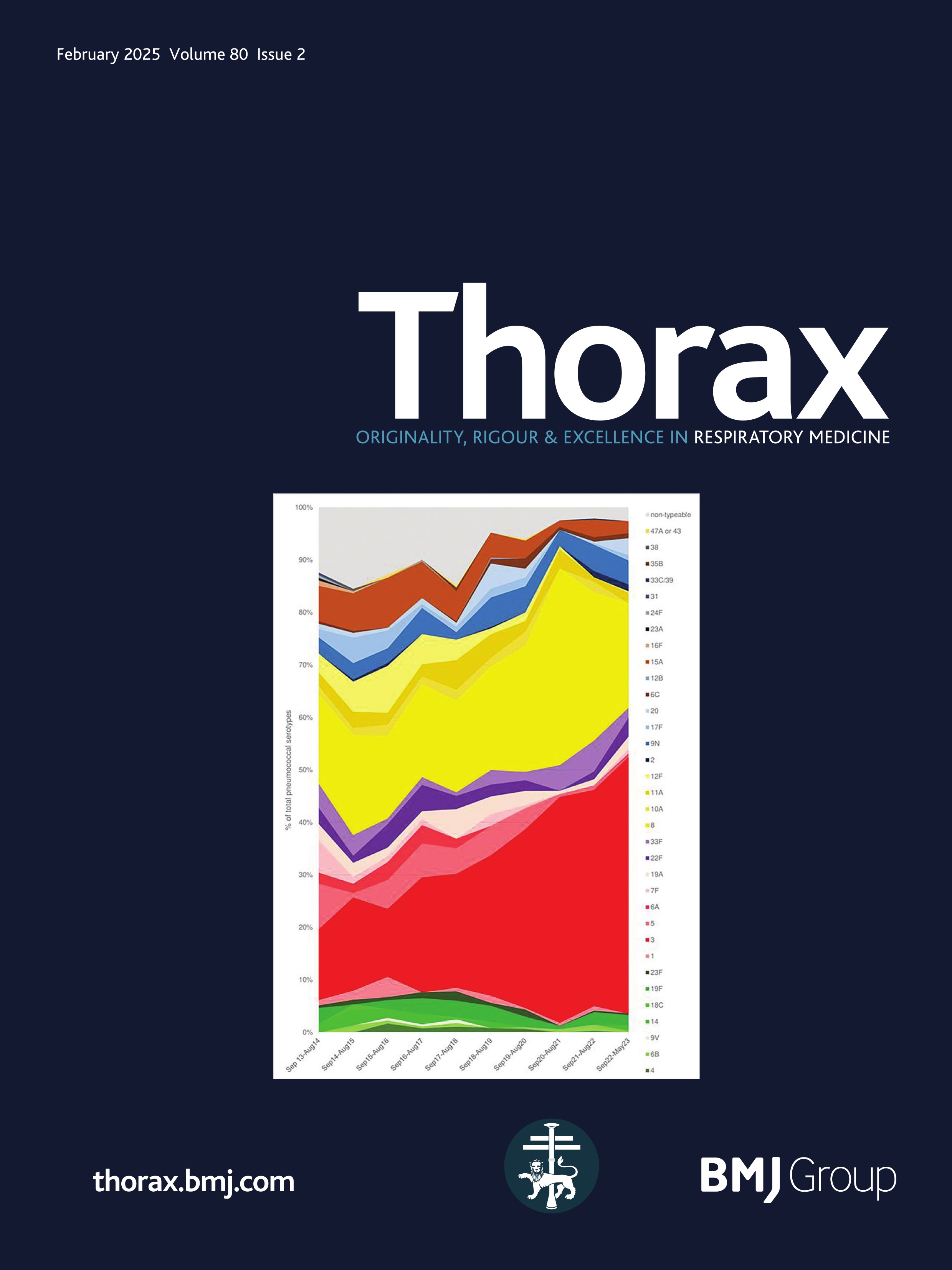
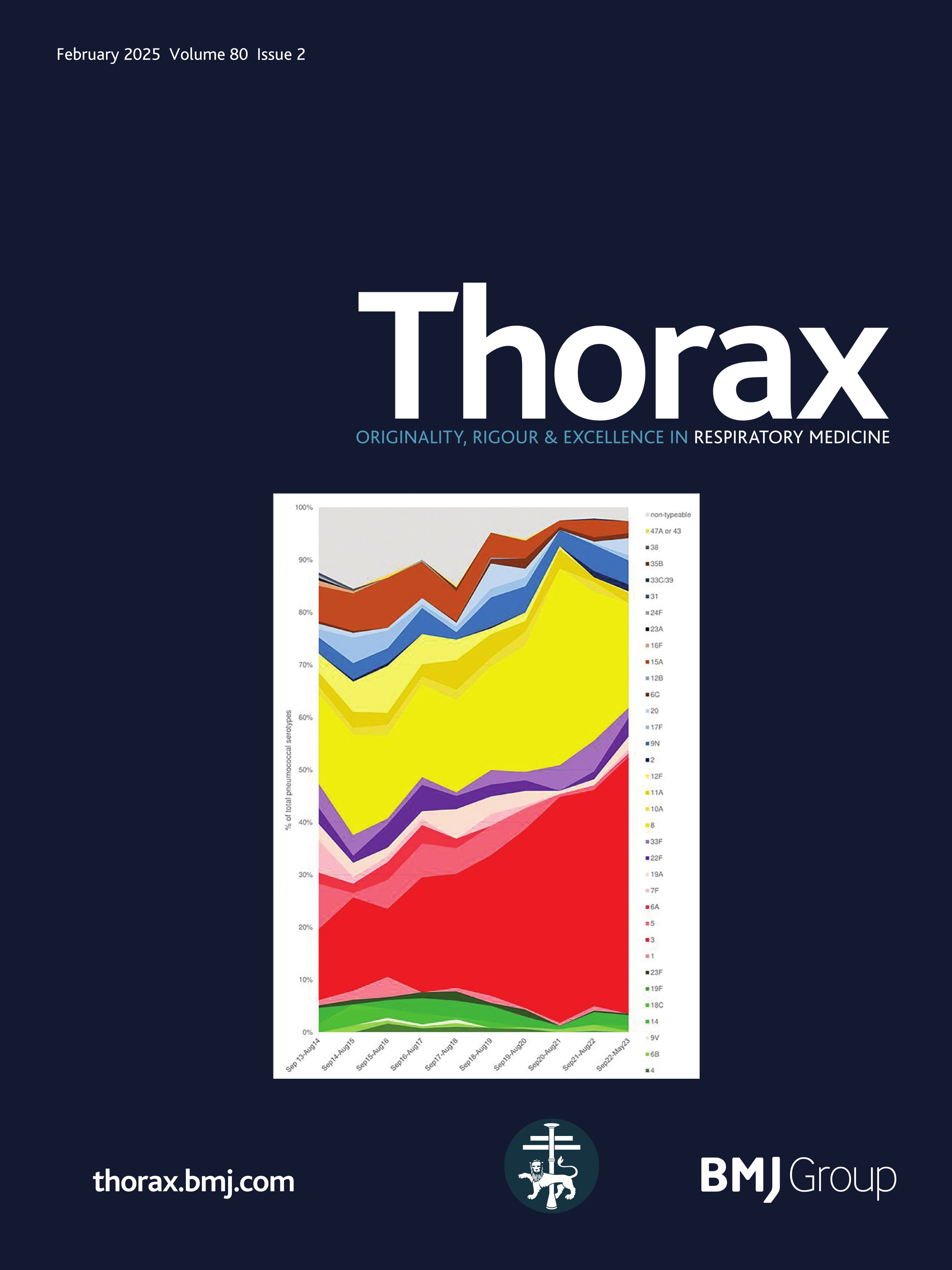
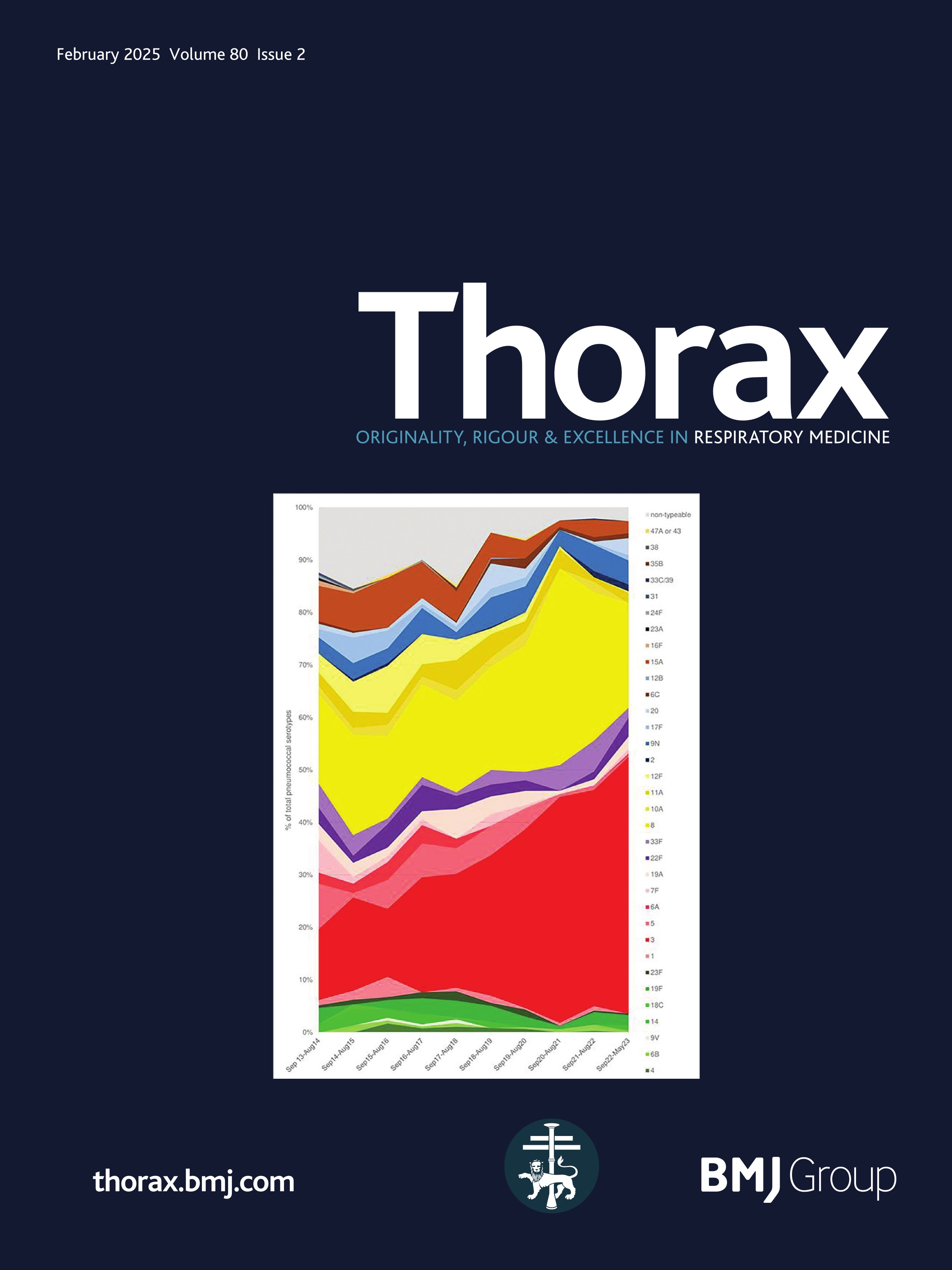
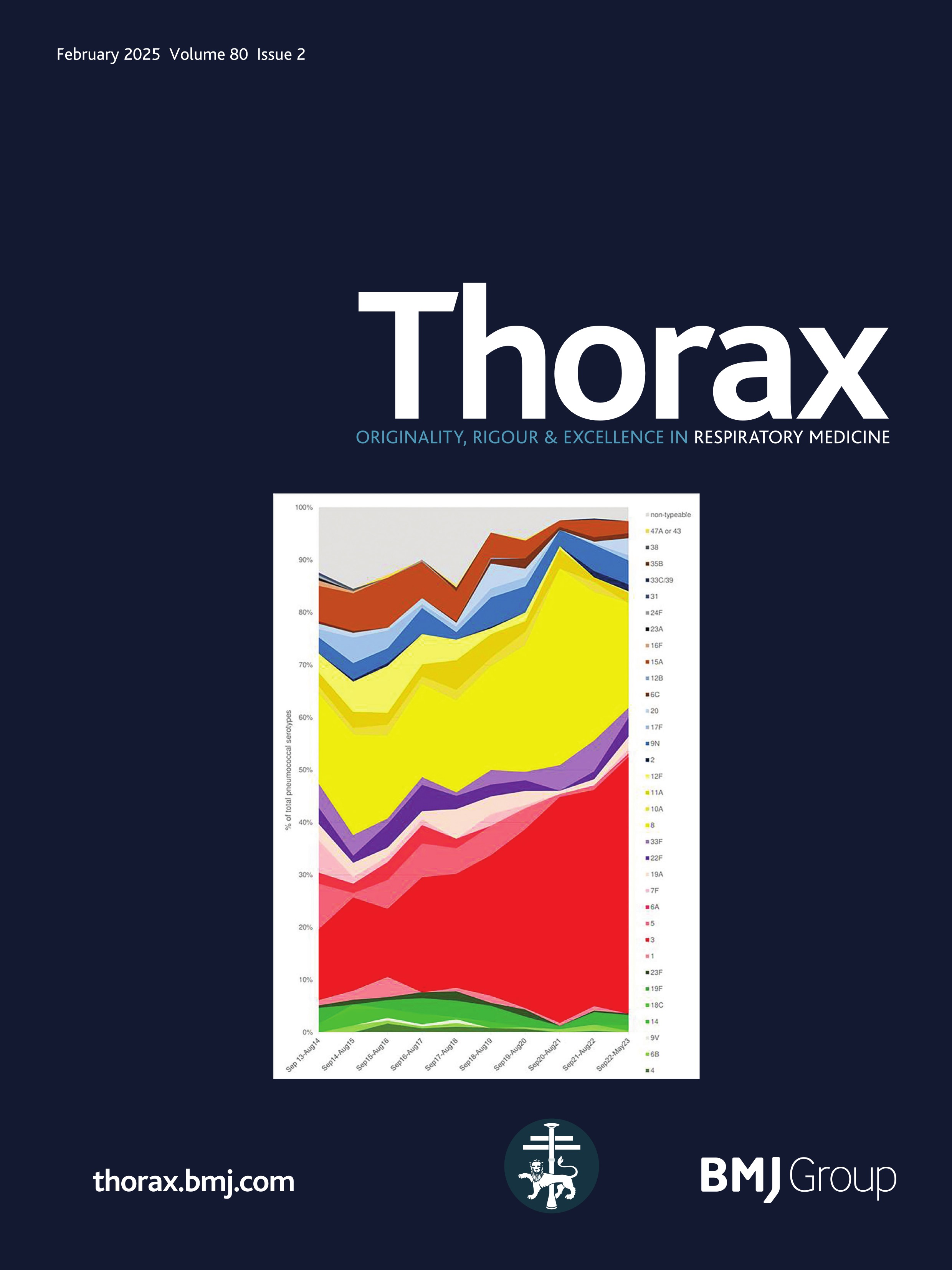
ResultsPresentationAmong the 11 identified patients from eight families, eight patients carried homozygous SFTPB variants and three were compound heterozygous (table 1, online supplemental table S1). The most common pathogenic variant p.(Pro121Glufs*95) was found in 6/11 (54.5%) of the patients in homozygous (n=3) or compound heterozygous (n=3) state. Three related paediatric patients with the previously reported hypomorphic homozygous splicing variant c.582G>A were considered separately.4 All other variants were frameshift or nonsense variants associated with a predicted absence of SP-B production (figure 1). The majority of the cohort presented symptoms at birth with a neonatal RDS in 10 patients (91%) including all the patients with loss-of-function mutations. One patient with the homozygous c.582G>A splice variant showed symptoms in the first month of life. None of the other patients presented after the neonatal period. At the onset, the disease was very severe: the Fan score reached 5/5 in 45% of the patients (n=5/11), indicating a PHT associated with respiratory failure, and 4/5 in 55% of the patients (n=6/11).5
Table 1Baseline and follow-up characteristics of patients with bi-allelic SFTPB mutations
 Figure 1
Figure 1 Domain organisation of SFTPB sequence variations observed in the cohort. Nomenclature follows the Human Genome Variation Society (HGVS) recommendations using the matched annotation (MANE) transcript as reference (NM_000542.5). The position of domains of SFTPB was referred to the prediction by SMART from the SP-B sequence, according to Ueno et al, J Biol Chem (2004). Mature protein (amino acids 201–279) is represented in dark blue. SapA and SapB, saposin A and B. Sp, signal peptide. # Splicing variants. The hypomorphic splicing variant c.582G>A is represented in blue and the other variants in red.
For 5/11 (45%) of patients, only chest X-rays were available and revealed a persistent bilateral alveolar syndrome. The 6 (55%) patients who were able to undergo CT imaging had diffuse or patchy GGO involving multiple lobes (figure 2). Echocardiography confirmed PHT in 5/8 (62%) SP-B-deficient patients and in none of the three patients with the hypomorphic variant.
 Figure 2
Figure 2 Main patterns seen on high-resolution axial CT and on lung biopsy histological analysis and survival of patients with SFTPB mutations. (A, B, C) 1-day-old to 15-day-old patients with widespread and dense GGO. (D) The 3-month-old patient carrying c.582G>A variants with a childhood interstitial lung disease since birth showing GGO. (E and F) Newborn patient with fatal SP-B deficiency. The lung parenchyma is dense and poorly aerated. The low magnification (E, ×50, scale bar 800 µm) shows a pattern of diffuse developmental disorder of the lung with features of alveolar capillary dysplasia without misalignment of pulmonary veins. At high magnification (F, ×200, scale bar 200 µm), thickened alveolar walls with hyperplastic pneumocytes can be seen together with alveolar distortion consistent with a fibrosing process. In this patient, a next-generation sequencing panel for the genes involved in PHT was negative. (G) Pro-SP-B staining (polyclonal antibody HPA062148, Sigma, 1/20 targeting the 121–197 amino acids) of patient 2, a newborn with the compound heterozygous variants c.111G>A, p.(Trp37*) and c.397delinsGAA p.(Pro133Glufs*95). (H) A complete loss of expression is observed at magnification ×400 (scale bar 100 µm) and ×1000 compared with an infant control. (I). Kaplan-Meier survival curve of patients with mutations in the SFTPB gene. Survival curves of the two genotype groups were compared using the log-rank test (p value=0.023).
No bronchoalveolar lavage (BAL) was available at birth. Diagnostic lung biopsy (n=3) or autopsy slides (n=4) were available for review in 7 (64%) patients and found patterns of non-specific interstitial pneumonia (n=1), pulmonary fibrosis (n=1), alveolar proteinosis (n=3) and diffuse developmental disorders (n=3) (online supplemental table S1). When performed, SP-B staining showed a complete absence of SP-B expression in two patients (17GM02316 & 18GM01191) and a normal to increased cytoplasmic SP-C staining in two patients (18GM01191 and 8824GM000165).
Treatments and outcomeAll patients with complete SP-B deficiency presented with severe neonatal respiratory distress requiring mechanical ventilation (n=8/8, 100%) (table 1). Conversely, none of the patients with the hypomorphic variant required mechanical ventilation. Exogenous surfactant was administered to 6/10 (60%) of the patients with a positive but transient improvement in oxygen requirement in all. The most common therapy given to patients (n=4) was systemic corticosteroids with no to poor benefit.
Survival curves differed significantly according to genotype (p=0.023). Except for the three patients with the c.582G>A variant, all the patients died at a median age of 1 month (figure 2I), PHT being an important prognostic factor (p=0.037). The ages at the last follow-up of the three relatives who survived after the neonatal period were 1, 11 and 21 years. Available lung function tests from the oldest patient found an FVC at 51% of predicted values and an FEV1 at 34%.
DiscussionThe present study describes for the first time a series of 11 paediatric patients with bi-allelic SFTPB variants. It highlights that complete SP-B deficiency is always lethal with no significant variation in disease expression. On the contrary, the study confirms that hypomorphic splice variants may preserve a variable proportion of normal SFTPB transcripts allowing survival.
In France, SP-B deficiency accounts for 1.3% of chILD and 10% of surfactant disorders.2 Unusual neonatal respiratory distress should lead to a quick CT scan. This study consistently showed dense and diffuse GGO and alveolar consolidations with no cystic formations, as observed in ABCA3 deficiency.6 Histology may show diffuse abnormal lung development, suggesting that SP-B plays a crucial role in distal bronchial and alveolar development during foetal life.7 This is further suggested by transgenic mice supporting that a critical level of SP-B is necessary for normal airspace formation and lung function.8 9 Genetic analyses confirmed the diagnosis. However, the low number of patients retrieved with SFTPB variants could be due to undiagnosed cases of rapid and unexplained neonatal death or to less severe forms with no or moderate neonatal respiratory signs. Additionally, SP-B deficiency may be related to molecular variations in genes involved in post-translational processing of SP-B, as recently shown for RAB5B.10
The most common p.(Pro121Glufs*95) variant, as well as the two first described nonsense variants p.(Trp25*) and p.(Tyr253*) result in a premature stop codon leading to the absence of mature SP-B and an abnormal alveolar development.3
Despite intensive management, none of the patients with an absence of SP-B survived beyond the neonatal period. Lung transplantation was reported with an overall prognosis similar to those of patients with other lung diseases but is not yet considered for newborns in France.11 12 Another promising therapeutic approach may come from neonatal gene therapy, an option that is currently at a preclinical stage.13
In conclusion, this study described the genotype and phenotype of chILD associated with pathogenic variants in SFTPB. The prognosis of patients with a genotype leading to the absence of SP-B appears to be fatal, whereas very rare cases of hypomorphic variants may allow survival (online supplemental table S2). In the case of unexplained severe RDS requiring invasive ventilation in a full-term child, genetic analyses for surfactant disorders should be undertaken urgently to guide the management and offer appropriate genetic counselling to the parents.
Ethics statementsPatient consent for publicationEthics approvalThis study involves human participants and was approved by the local ethics evaluation committee of the French pulmonology society (Société de Pneumologie de langue française SPLF CEPRO, reference n°2022-054). Data collection has been validated by legal authorities (Comité National de l’Informatique et des Libertés, CNIL n°08.015bis) and by an ethical committee (Comité consultatif sur le Traitement de l’Information en matière de Recherche dans le domaine de la Santé, CCTIRS, 20080328). Participants gave informed consent to participate in the study before taking part.
AcknowledgmentsWe thank the Assistance Publique-Hôpitaux de Paris (APHP), Sorbonne Université (SU), Paris, France, and the Institut National de la Santé et de la recherche Médicale (Inserm). We thank the national networks for rare lung diseases: Centre de référence des maladies respiratoires rares (RespiRare, www.respirare.fr) and Filière de santé pour les maladies respiratoires rares (RespiFIL, www.respifil.fr) and the Rare diseases Cohort project for ILD (RaDiCo-PID). We thank the Banque National de Données Maladies Rares (BNDMR) for their collaboration. The chILD studies are part of the European Respiratory Society (ERS) Clinical Research Collaboration for chILD (CRC chILDEU), with the support of the European Lung Foundation (ELF) chILD group.
Comments (0)