
Remember me
Antibiotics are a class of compounds initially used for the treatment and prevention of bacterial infections. Unfortunately, with the overuse and misuse of antibiotics, bacteria begin to develop the ability to defeat them, leading to the emergence of antimicrobial resistance (AMR) and antibiotic-resistant bacteria (ARB) [1,2,3]. At present, the problem of AMR is becoming increasingly serious worldwide and has become one of the major causes of disease and death, causing a serious global health crisis. According to statistics by The Lancet, antibiotic-resistant infections killed 1.27 million people in 2019 alone [4], and are responsible for roughly 5 million deaths in 2022 [5]. It is estimated that by 2050, this number will increase to 10 million people per year, significantly exceeding the number of cancer deaths (8.2 million) [5, 6]. Moreover, AMR also poses a significant burden on healthcare systems, resulting in high global health expenditures. Based on the US Centers for Disease Control and Prevention (CDC) data, antibiotic-resistant infections are estimated to cost up to $20 billion in annual healthcare costs and $35 billion in lost productivity [7]. To combat the growing threat of AMR to human health and the economy, understanding the underlying molecular mechanisms is crucial for developing new strategies to treat infectious diseases.
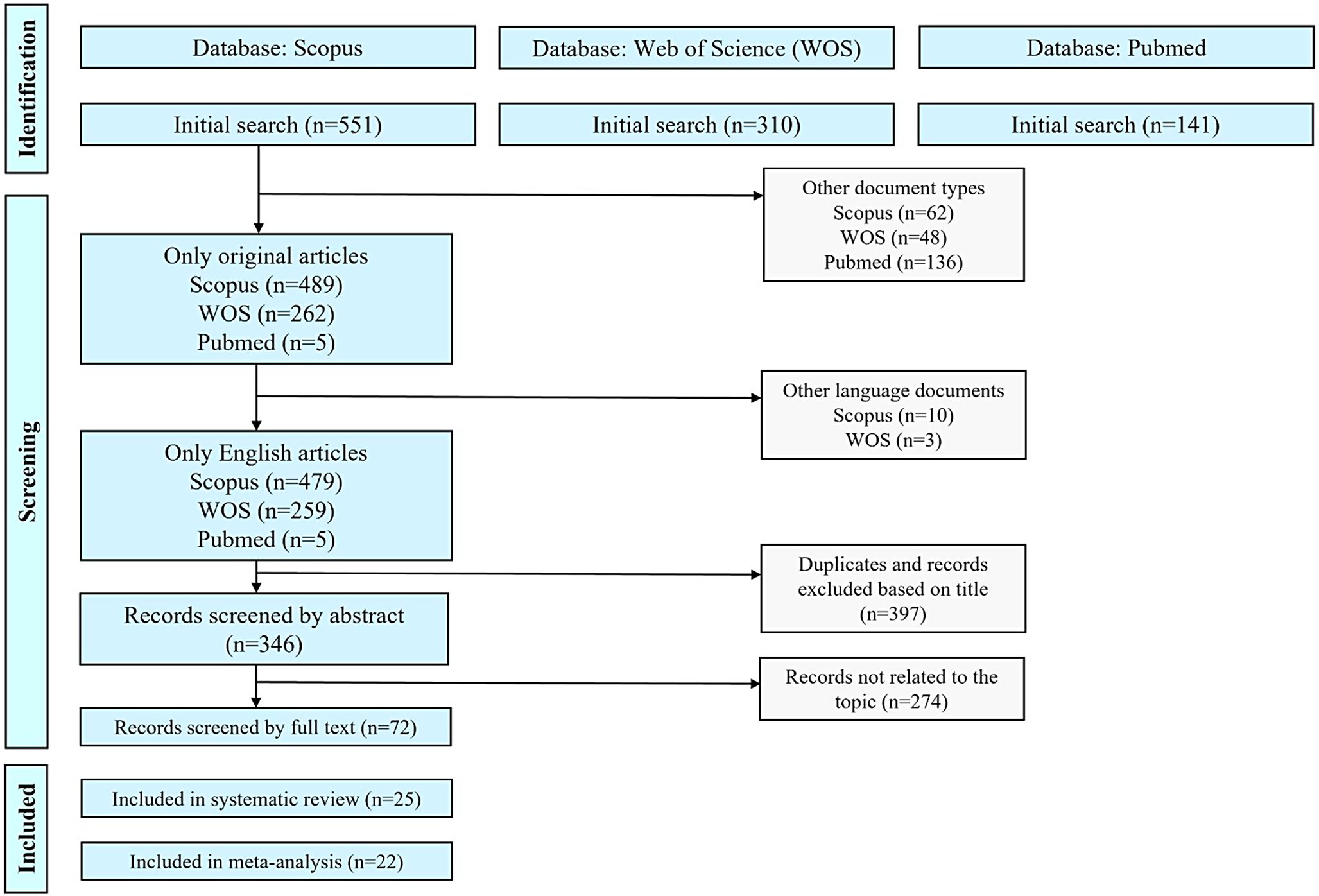
An increasing number of studies are revealing the mechanisms of action of antibiotics and the AMR mechanisms in environmental microbes (Fig. 1). The antibiotics usually share the same ecological niche with bacteria, and it is generally believed that growth inhibition is their main ecological function in the environment. There are several essential antibiotic targets in bacteria (e.g., cell wall, cell membrane, nucleic acid, and ribosome) [8, 9]. Accordingly, the antibiotic action mode includes (1) inhibition of peptidoglycan synthesis in the process of forming the cell wall; (2) interfering with cell membrane function through the eventual disruption of the outer and cytoplasmic membrane; (3) inhibition of nucleic acid synthesis through inhibiting bacterial RNA polymerase activity and blocking transcription; (4) targeting protein synthesis. Unfortunately, in order to survive, bacteria retain an outstanding capacity for rapid adaptation and evolution in response to various antibiotics through various molecular mechanisms [10].
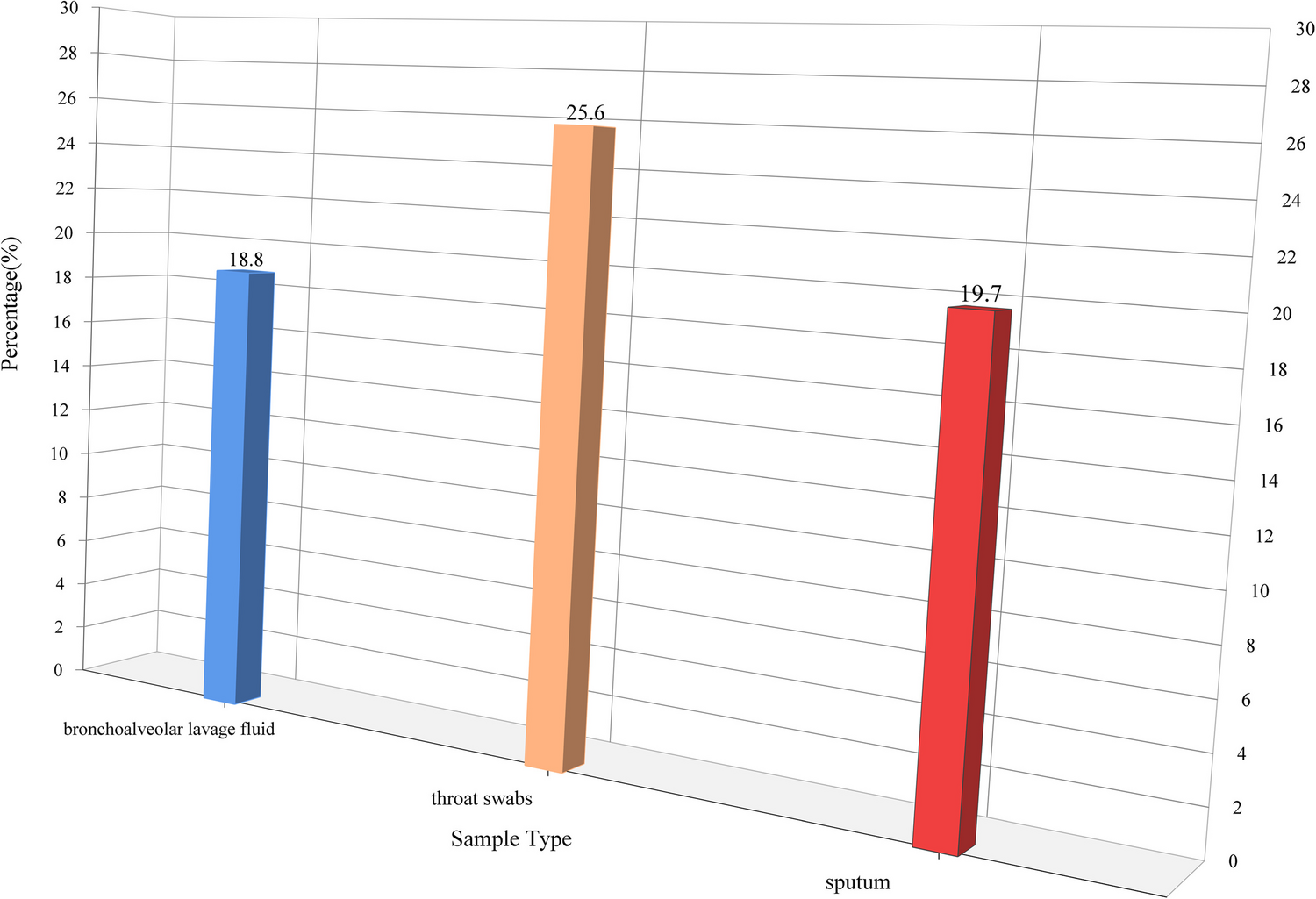
Fig. 1
General mechanisms for antibiotic action and antimicrobial resistance. The antibacterial activity of antibiotics relies on four main mechanisms: (1) inhibition of peptidoglycan synthesis; (2) disruption of the cell membrane; (3) inhibition of nucleic acid synthesis; and (4) interfering with protein synthesis [9]. For typical AMR mechanisms, efflux pump, inactivation of antibiotics by modifying enzymes, alteration of membrane permeability, modification of antibiotic target, and target protection are included [11]
Bacteria can be intrinsically resistant to one or more classes of antibiotics. They can also acquire such resistance by either (1) genetic mutations in the chromosome or (2) acquired exogenous DNA via horizontal gene transfer encoding resistance determinants [12]. A number of genes that confer intrinsic resistance to different kinds of antibiotics (such as β-lactams, fluoroquinolones, and aminoglycosides) have been identified in the genomes of bacteria [11, 13]. Mutations are a permanent change for bacteria to become resistant to antibiotics, which typically involve a few types of genes, including those encoding the antibiotic targets, antibiotic transporters, and regulators that modulate the expression of transporters, such as efflux pumps and modifying enzymes, thereby causing AMR [14]. Acquired antibiotic resistance is the result of an evolutionary process where bacteria are endowed with various mechanisms, such as active efflux of the antibiotic, enzymatic modification/degradation of the antibiotic, alteration of membrane permeability, modification of antibiotic targets, and target protection [15, 16]. These sophisticated mechanisms have been well-documented in previous studies [11, 17,18,19] and significantly contributed to the development and clinical use of many AMR breakers, such as antibiotic resistance gene (ARG) silencers, ribosomal inhibitors, and efflux pump inhibitors [20]. However, AMR is a naturally occurring process that will never go away [21, 22]. Bacteria, especially those prevalent pathogens (e.g., methicillin-resistant Staphylococcus aureus, penicillin and β-lactam-resistant Streptococcus pneumoniae, pan-resistant Acinetobacter baumannii, and multidrug-resistant Pseudomonas aeruginosa) are always evolving new ways to avoid the effects of the antibiotic [23]. Hence, the continued fight against bacterial infections/resistance to antibiotics is required.
Over the past years, new technological advances have driven continuous progress in revealing the molecular mechanisms of AMR. The underlying resistance mechanisms contribute to our understanding of the evolution of AMR in bacteria. More importantly, clarifying these mechanisms is crucial for improving antibiotic efficacy, obtaining answers, and exploring options to combat bacterial resistance to preserve the utility of antibiotics for years to come. In these cases, following the five general mechanisms (Fig. 1), this review summarizes recent progress in understanding AMR by compiling emerging evidence from previous studies (2020–2025), which may offer insights into the development of a novel approach to overcome AMR in environmental and clinical settings.
Efflux pump-like proteinsEfflux pumps play a prominent role in controlling the accumulation and transportation of antibiotics in resistant bacteria. It is the most studied and targeted AMR mechanism. The first efflux pump expelling tetracycline in Escherichia coli was discovered in 1980 [24]. Since then, various efflux mechanisms have been described to account for bacterial infections. According to the substrate selectivity, protein structure, and energy source, several well-known antibiotic efflux pump families have been identified, including the major facilitator superfamily (MFS), the ATP-binding cassette (ABC) superfamily, the drug metabolite transporter (DMT) superfamily, the small multidrug resistance (SMR) family, the resistance-nodulation-division (RND) superfamily, the multidrug and toxic compound extrusion (MATE) family, and the proteobacterial antimicrobial compound efflux (PACE) superfamily [25]. Most plasmid-carried or chromosomal genes encoding efflux pumps among gram-positive bacteria belong to the MFS and ABC families. In contrast, the predominant clinically relevant efflux system in gram-negative bacteria usually comes from a member of the RND superfamily, comprising an inner-membrane protein, periplasmic adapter proteins, and outer-membrane protein. Accumulating evidence demonstrates that the efflux pumps are widely implicated in (a) efflux of antibiotics, (b) regulation of host physiology, (c) biofilm formation, (d) metal resistance, and (e) virulence [26], suggesting they are more clinically important than usually thought. Recently, there has been an increasing number of proteins with functions similar to efflux pumps. These efflux pump-like proteins have undeniable significance in intrinsic and acquired resistance to various antibiotics in bacteria. Some reported examples are shown below.
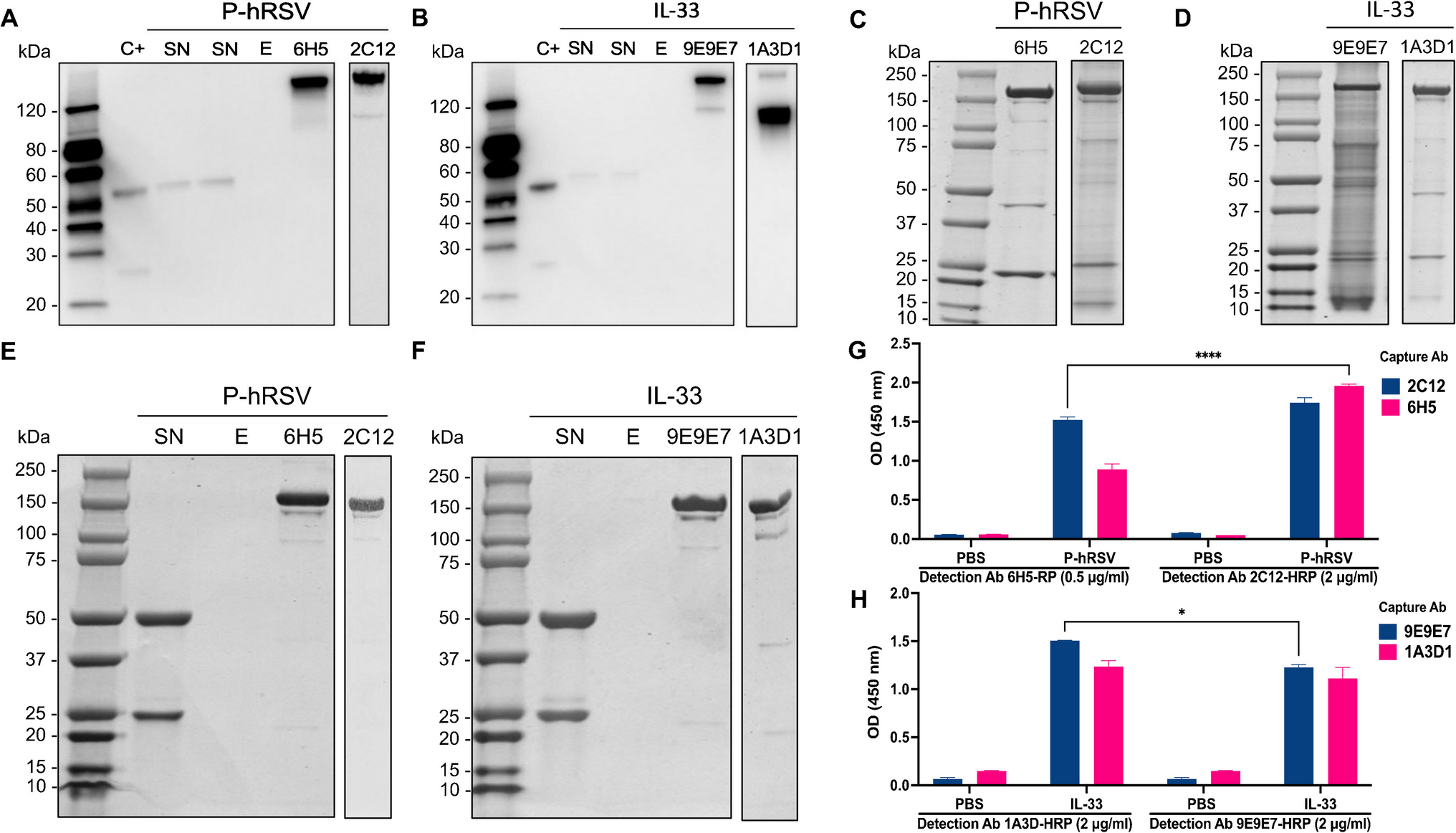
BON domain-containing proteinEfflux pumps have been identified as the dominant resistance mechanism in nearly all organisms, while other cytoplasmic and membrane proteins have received little attention. Nevertheless, they play an equally important role in promoting resistance to infections. These intrinsic resistances in many bacteria represent even more serious therapeutic problems. Among them, the BON (bacterial OsmY and nodulation), a putative membrane-binding domain, is one of the most abundant proteins in the cell membrane [27]. It has been demonstrated to bind noncovalently to peptidoglycan and chitin through the interaction with N-acetyl glucosamine moieties [28]. A recent study has suggested that the BON domain-containing protein exhibits a high affinity toward carbapenem antibiotics [29]. The bacteria containing BON protein showed fold increases in higher minimum inhibitory concentration (MIC) values of imipenem and meropenem in comparison to the NP-6 clinical strain and E. coli DH5α. Molecular dynamic simulation studies revealed that BON protein can form stable complexes with meropenem and imipenem. Subsequently, Sun et al. reported another new type of BON domain-containing protein with an efflux pump-like function which was identified from the soil metagenomic libraries [30]. It helped bacteria resist diverse antibiotics, with the highest efflux activity toward ceftazidime. A more than 32-fold increase in MIC was observed. Moreover, the BON protein was active in the co-selection of antibiotic and metal ion resistance in the bacterial cells. Structurally, the BON protein can undergo self-assembly into a trimer complex, forming a pore-shaped channel to transport antibiotics. A conservative WXG motif in the BON protein is essential for the substrate-transporting function, formation of oligomeric pores, and interactions between proteins and cell membranes. Based on these findings, an AMR mechanism, namely “one-in, one-out”, was proposed for the first time (Fig. 2a). Further studies are required to shed light on the sequence-structure-function relationships in the BON domain-containing protein. For example, exploring the sequence and structural diversity, determining the crystal structure, and understanding the resistance mechanism would fill the knowledge gap of BON domain-containing protein-mediated antibiotic resistance.
Fig. 2
Emerging efflux pump-like proteins-induced antibiotic resistance mechanisms. (a) BON domain-containing protein-mediated co-selection of antibiotic and heavy metal resistance in bacteria [30]. (b) Cryo-electron microscopy structures of a Campylobacter multidrug efflux pump CmeABC without antibiotics [31]. Each subunit of CmeB is labelled with a different colour. The six subdomains DN, DC, PN1, PN2, PC1, and PC2 are labelled. (c) A NiCoT family metal transporter from Mycobacterium tuberculosis (Rv2856/NicT) behaves as a drug efflux pump facilitating cross-resistance to antibiotics [32]. All the antibiotics in the active site are shown after docking. The docking interaction of NicT with the gentamicin on the right side is used as an example. Several key amino acid residues are labelled. (d) Bacitracin resistance and enhanced virulence of Streptococcus suis via a novel membrane transporter SstFEG [33]
CmeABC multidrug efflux systemCmeABC multidrug efflux system consists of three tripartite protein components, including CmeA, CmeB, and CmeC, which play a central role in mediating multidrug resistance to many antibiotics such as chloramphenicols, fluoroquinolones, tetracycline, and macrolides in Campylobacter jejuni (C. jejuni) [34]. CmeB contains the specific substrate binding sites involved in the proton-relay network that are responsible for the proton motive force-dependent active transport. CmeA is a periplasmic membrane fusion protein, while CmeC forms an outer membrane channel. Three components play a synergistic role in antibiotic expulsion [35]. Recently, Yao et al. identified a potent variant of CmeABC, namely RE-CmeABC, which significantly enhanced Campylobacter’s resistance to multiple antibiotics [36]. It was found that the C. jejuni strain carrying the RE-CmeABC expanded mutational selectivity to ciprofloxacin, increased the frequency of mutation to ciprofloxacin resistance, and altered the MIC distributions of several drugs in clinical C. jejuni isolates to a higher range. Consequently, the RE-CmeABC efflux system is considered an effective strategy utilized by Campylobacter for adaptation to antibiotic selection, representing an emerging multidrug-resistant mechanism [36]. To elucidate the molecular mechanisms underlying antibiotic resistance, Zhang et al. determined the 3D structures of this membrane protein RE-CmeB in complex with some antibiotics such as chloramphenicol, ciprofloxacin, ampicillin, and erythromycin [31] (Fig. 2b). Based on these 3D structures, key ligand-binding residues as well as important interactions between RE-CmeB and antibiotics were identified. It suggested that different subsets of amino acid residues in RE-CmeB protein were involved in binding the antibiotics, which contributes to the optimization of the substrate identification patterns and a strong capability to extrude a broad spectrum of antibiotics effectively. Further structural investigations are needed to explain the detailed molecular mechanisms of RE-CmeB transporter controlling multidrug recognition. How RE-CmeB transports antibiotics out of cells and which amino acid residues play important roles in this process are unanswered scientific questions.
NiCoT transporterMetal ions often serve as co-selecting factors in the proliferation of AMR in human pathogens from given environmental settings. The resulting enhanced antibiotic transport typically results in low levels of intrinsic susceptibility, acquisition of collateral resistance mechanisms, and cross-resistance to chemically unrelated compounds. For example, the NiCoT transporter family is one of the most widespread Ni/Co transporters in various organisms [37]. Recently, Adhikary et al. reported the ability of a putative NiCoT family transporter, Rv2856 or NicT from Mycobacterium tuberculosis (Mtb), to transport nickel/cobalt and antibiotics and identified a group of key amino acid residues responsible for its function [32]. The increase of NicT-induced intracellular nickel uptake in E. coli CS109 and Mycobacterium smegmatis resulted in enhanced resistance to several antibiotics such as ofloxacin, gentamicin, norfloxacin, sparfloxacin, isoniazid and nalidixic acid. Compared with cells without NicT, intracellular accumulation of norfloxacin was relatively low when NicT was expressed in the bacterial cells, suggesting that NicT is involved in the active process of antibiotic efflux. Furthermore, results of the docking study revealed that a number of conserved residues such as Asp82, His83, and Asp88 in domain II of NicT contributed to the formation of the H-bond networks (Fig. 2c). The residues Ala81, Ile84, His112, Phe222, Asp227, Thr228 and Thr230 were the major contributors to hydrophobic and van der Waals interactions. The study demonstrated that Rv2856/NicT was able to actively export different antibiotics and yield cross-resistance to certain antibiotics in the presence of nickel.
AadT pumpIn clinical settings, multidrug efflux pumps have been described in the development of Acinetobacter strains’ resistance to multiple antibiotics [38]. The genes encoding putative pumps are mostly identified in the core genome of the species [39]. Others are implicated with the concerted activities of mobile genetic elements (e.g., transposons and plasmids), dispersed in a subset of phylogenetically distant bacterial strains [40]. For instance, the tetracycline efflux pump, TetB, which was initially discovered on the transposon Tn10 in E. coli, was reported in many Acinetobacter strains and other gram-negative species [41]. Recently, a group of genes encoding an efflux pump belonging to the Drug: H + antiporter 2 (DHA2) family were identified in several plasmids of Acinetobacter [42]. The novel DHA2 family pump proteins can reduce susceptibility to a variety of antibiotics. Moreover, the genes encoding pump homologs are prevalent but invariably related to variants at the AdeAB(C) locus in the corresponding Acinetobacter species. Among them, AadT, a new efflux pump protein, was found in the plasmids of Acinetobacter’s multidrug resistance [43]. It showed a strong ability to reduce bacterial susceptibility to several antimicrobials, such as erythromycin, tetracycline, and chlorhexidine. The AadT homologs are usually adjacent to the AdeAB(C) efflux pump variants in many Acinetobacter species, suggesting that AadT might be able to cooperate with AdeAB(C) variants in the Acinetobacter resistance arsenal. More research data is required to confirm the relationships between AddT and other efflux pumps in the Acinetobacter species.
Novel membrane transporter-SstFEGStreptococcus suis is among the top zoonotic pathogens that cause diseases in pigs and humans, representing a global health problem in the swine industry [44]. It is widely present in the breeding environment of pigs and is considered a reservoir for antibiotic resistance genes [45, 46]. Bacitracin is a polypeptide antibiotic used extensively as a growth promoter in animal husbandry [47]. However, long-time use of bacitracin leads to an increase in resistance genes in microbial strains. Several bacitracin resistance mechanisms have been uncovered over the last decade [48, 49]. For example, the Bce systems composed of the regulator BceSR and the transporter BceAB are widely distributed in bacteria and have been identified as an efflux pump conferring resistance to bacitracin [50]. More recently, a study showed that a potential efflux pump SstFEG is located upstream of the known bacitracin resistance genes bceAB and bceRS [33] (Fig. 2d). The deletion of sstFEG significantly decreased the mutant susceptibility to bacitracin in comparison to wildtype S. suis strain. Furthermore, it was found that both the BceAB transporter and the two-component system BceRS were required for SstFEG-mediated bacitracin resistance. Additionally, by dissecting the competitive survival advantage of S. suis in animal infection, SstFEG played an important role in colonization and virulence.
SA09310 proteinStaphylococcus aureus is a gram-positive pathogen causing a range of clinical diseases [51]. S. aureus is a notorious human pathogen partly due to its capacity to survive and fight against a variety of antimicrobial compounds [52]. S. aureus has evolved various antibiotic resistance mechanisms in the treatment [53, 54]. Among them, efflux pump-mediated resistances in S. aureus have been the most extensively studied [55]. According to in silico analysis, 31 multidrug efflux pumps in the S. aureus chromosome cover almost all the pump protein families. Nevertheless, only one-third of them have been previously characterized, while the biological functions of these efflux pumps remain poorly understood [56]. In a recent study, a novel multidrug resistance efflux protein, namely SA09310, encoded in the chromosomes of S. aureus, was identified and characterized [57], which consisted of 12 transmembrane helices. A sa09310 gene knockout mutant (Δsa09310) showed enhanced sensitivity to doxycycline and tetracycline, with the MICs increased by 8-fold and 64-fold, respectively. It was demonstrated that the SA09310 possessed the strong capacity to export tetracycline from intracellular to extracellular space. The high conservation of SA09310 homologs in Staphylococcus suggests that protein clusters similar to the SA09310 have general functions. More studies are required to decode the regulatory mechanism of AMR caused by SA09310.
Inactivation of antibioticsSome bacteria acquire AMR by enzymatically degrading or modifying antibiotic molecules to render them inactive and potentially protect neighboring susceptible cells from antibiotic exposure, which is increasingly prevalent among pathogens. For example, the β-lactamases are a well-known example of antibiotic degradation enzymes, which are mostly produced by the enterobacterales and some species of gram-positive bacteria (e.g., Enterococcus faecalis, S. aureus, and Enterococcus faecium [58]. Based on the protein sequences, they can be separated into four distinct classes (A, B, C, and D) and are highly efficient in inactivating commonly used β-lactam antibiotics by breaking the β-lactam ring open, making them ineffective in binding with the target [59]. In addition, the transfer of chemical groups such as phosphoryl, acetyl, and adenyl is the most common pathway of antibiotic inactivation. A number of transferases (such as N-acetyl transferases, O-phosphotransferases, and O-adenyltransferases) have been identified and characterized for their ability to transfer these chemical groups. Phosphorylation and adenylation are the primary approaches rendering aminoglycosides inactive, while acetylation is widely utilized to inactivate streptogramins, fluoroquinolones, and chloramphenicol [15]. More recently, several studies have reported that oxidative and reductive inactivation of antibiotics often leads to novel resistance mechanisms. Expanding our understanding of these emerging AMR mechanisms is of crucial importance to combat the AMR crisis in the world.
Oxidative inactivationChloramphenicols have become the main focus of research because of the absence of new antibiotic formulations and the emergence of AMR resulting from the overuse and misuse of existing antibiotics. Chloramphenicol (CAP), thiamphenicol (TAP), and florfenicol (FF) are synthetic antibiotics commonly used in the treatment of infections due to their broad range of activity against bacteria and low cost. CAP contains a p-nitrophenyl group, a dichloromethyl moiety, and a propanediol side chain [60]. The accumulation and distribution of chloramphenicols have been recorded in hot spots such as pharmaceuticals, livestock, poultry, and hospital wastewater. They are the main reasons for the development of chloramphenicols-resistant bacteria. Several chloramphenicols resistance mechanisms have been reported, such as enzymatic inactivation [11]. For example, the acetyltransferases-catalyzed acetylation of the propanediol group of CAP and TAP has been frequently recognized in many bacteria [61]. The hydrolysis of amide bonds by hydrolase estDL136 has also been connected to the fast inactivation of CAP, TAP, and FF [62]. In a previous study, Zhang et al. showed a CAP-resistant strain Sphingomonas sp. CL5.1 possessed the ability to oxidize the propanediol C3-OH groups of CAP and TAP into carboxyl groups [63] (Fig. 3a), which is a necessary pharmacophore for the CAP and TAP antibacterial activities [64]. This finding represents a novel CAP resistance mechanism in bacterial cells. Subsequently, Zhang et al. discovered a novel oxidoreductase named CapO featuring the oxidative inactivation of CAP and TAP [65]. Ma et al. also reported a novel oxidase gene, cmO, identified from Sphingomonadaceae [66]. The corresponding oxidase CmO can catalyze the oxidation at the C-1’ and C-3’ positions of CAP and TAP in Sphingobium sp. strain. Docking studies showed that CAP was located within the active pocket of CmO through hydrogen-bonding interaction at key amino acid residues, such as G99, Y380, M474, and N518. Zhang et al. further reported the discovery and characterization of an oxidoreductase that inactivates CAP through dual oxidation of the C3 hydroxyl group [67]. Moreover, oxidation of CAP either relies on glucose-methanol-choline-type flavoenzymes alone, or on extra aldehyde dehydrogenases for increased efficiency. Overall, these results significantly expand our knowledge boundary about chloramphenicols resistance mechanisms. Additionally, resistance to rifamycin antibiotics known for their use in treating tuberculosis, has been revealed ranging from primary target modification and antibiotic inactivation to cytoplasmic exclusion [68]. Among them, rifamycin monooxygenases (Rox) are present in a variety of environmental bacteria and have been associated with the decomposition of the antibiotic. For example, Heba et al. showed that the recombinant rifampicin monooxygenase (RifMO) was able to catalyze the incorporation of a single oxygen atom forming an unstable intermediate that eventually is converted to 2′-N-hydroxy-4-oxo-Rif, leading to the rifampicin resistance in Nocardia farcinica [68]. Kalinka et al. reported an unprecedented mechanism of rifamycin inactivation initiated by monooxygenation of the 2-position of the naphthyl group, which subsequently resulted in ring opening and linearization of the antibiotic [69]. This represents a unique mechanism of enzymatic inactivation underpinning the broad spectrum of rifamycin resistance.
Fig. 3
Comments (0)