
Remember me

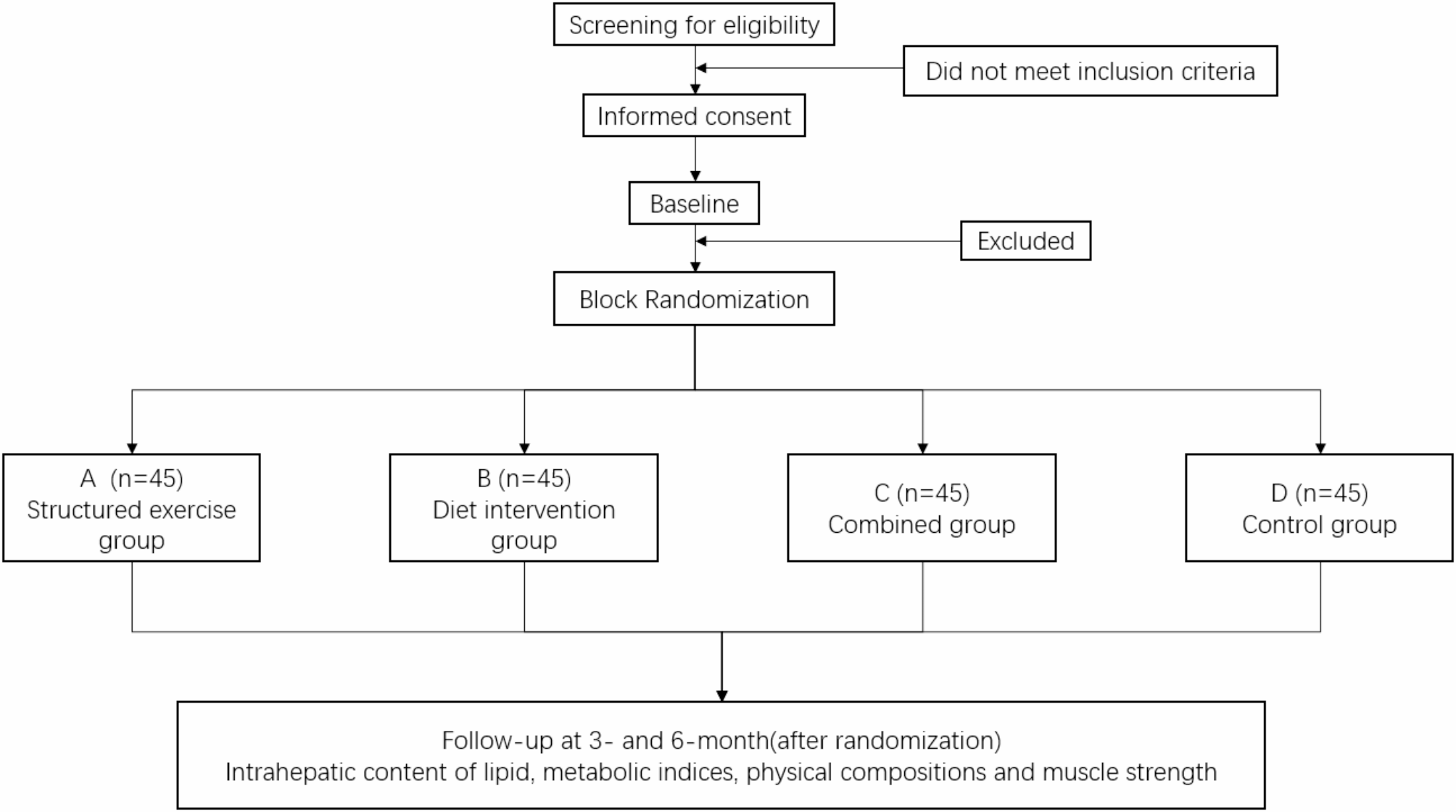





The study will be conducted at the Translation Medicine Research Center for Endocrine and Metabolic Disease of Beijing Friendship Hospital Pinggu Campus, Capital Medical University. This is a two-by-two factorial randomized controlled study to evaluate the efficacy of structured exercise and oat supplementation in NAFLD patients. Eligible participants with NAFLD will be randomly assigned (1:1:1:1) to the structured exercise group (aerobic exercise and resistance training), diet intervention group (80 g oats/daily supplementation), combined group (structured exercise + diet intervention) or control group by block randomization, and receive a corresponding intervention for 24 weeks. Intrahepatic lipid content, metabolic indices, physical composition and muscle strength will be evaluated at 12 and 24 weeks after start of intervention. A flow chart outlining participant flow is shown in Fig. 1.
This manuscript is based on Clinical Trial Protocol version 2.1 (June 25, 2021). The study was initiated in June 2021 and is expected to complete by the end of 2024. At the time of manuscript submission, a total of 156 participants have been enrolled in the study.
Fig. 1 Ethical approval
Ethical approvalThis study protocol and informed consent form have been approved by the Ethics Committee of Beijing Pinggu Hospital (Reference No: 2019 - Capital’s Funds for Health Improvement and Research 004 − 01).
ParticipantsParticipants will be recruited from the outpatient registration pool which includes those who have been diagnosed with NAFLD at the Endocrinology Department of Beijing Friendship Hospital Pinggu Campus, Capital Medical University. Participants who were diagnosed with NAFLD in our previous cohort study (Pinggu Metabolic Disease Study [8]) will also be invited to participate in this study. Potential participants will sign an informed consent form after we provide detailed information on the benefits and risks of this study, as well as measures for data and sample collection, storage and future use.
Inclusion criteriaThe inclusion criteria for the present study are as follows: (1) diagnosed with NAFLD (intrahepatic lipid content ≥5% according to quantitative computed tomography [21]) according to the World Gastroenterology Organization (WGO) global guidelines for NAFLD and non-alcoholic steatohepatitis (published in July 2014); (2) between the ages 18 and 65 years; and (3) willingness to participate in the study and signed informed consent.
Exclusion criteriaThe exclusion criteria include the following: (1) chronic liver disease (viral, autoimmune, or drug-induced injury) or serious impairment of liver function (≥2 times the normal upper limit of alanine aminotransferase/aspartate aminotransferase); (2) severe comorbidities such as myocardial infarction, uncontrolled hypertension (defined as systolic blood pressure > 140 mmHg and/or diastolic blood pressure ≥ 90 mmHg), chronic heart failure (New York Heart Association grade III/IV), chronic renal insufficiency (creatinine elevation), unstable or severe angina; (3) poorly controlled type 2 diabetes (haemoglobin A1C > 9.0% or fasting plasma glucose > 11.1 mmol/L), a diabetes course > 5 years or with severe complications, and other types of diabetes; (4) participation in a weight loss study within 3 months, participation in other clinical trials within one month; (5) chronic diarrhoea, history of intestinal diseases (such as Roemheld syndrome, severe hernia, intestinal obstruction, postoperative intestinal surgery and intestinal ulcer) or diseases that may worsen due to flatulence; and (6) excessive alcohol consumption (defined as weekly alcohol consumption > 210 g for men and > 140 g for women in the past 6 months); and (7) any other patients considered unsuitable by the researchers (including those with chronic consumptive diseases such as malignant tumour, tuberculosis, haematological diseases, mental diseases, autoimmune diseases, acute cerebrovascular diseases within the past 6 months, and those who are not suitable for exercise, such as those with limb disorders, or bone and joint diseases); (8) women with fertility (18–50 years old or less than 1 year after menopause) who are positive for human chorionic gonadotrophin at baseline, or are breast-feeding, unwilling to use appropriate contraceptives; (9) those who are allergic to ingredients used in the diet intervention; (10) individuals who have abused drugs in the previous 2 years; (11) those who have used weight-loss substances, antibiotics or intestinal flora regulator in the previous 4 weeks; (12) those who are not suitable for exercise intervention, according to the Physical Activity Readiness Questionnaire [22].
Early withdrawalParticipants or the legally authorized representatives have the right to withdraw from the study at any time. Participants who cannot tolerate the intervention and are deemed unsuitable by the attending physician to continue the study will also be withdrawn from the study. For participants who withdraw early from the study, the reasons for their early withdrawal and the last time point at which they received the intervention will be recorded. It is recommended that the study assessment items be completed for early withdrawal during the last visit to the extent possible.
RandomizationEligible participants will be randomized by block randomization. The random allocation codes will be generated by statistical professionals programmed on a computer using SAS 9.3 software. Given the number of seeds and the length of the block, a random grouping arrangement of 180 subjects will be generated according to the ratio of 1:1:1:1; that is, the intervention allocation corresponding to the serial number 001-180 will be listed, which is matched to the subject numbers, and will be printed and sealed in the envelope. Once the participants are selected and assigned numbers, the corresponding sealed envelopes will be opened. Participants will be randomly assigned in a 1:1:1:1 ratio to the structured exercise group, diet intervention group, combined group, or control group.
BlindingBlinding of the study participants and investigators is not possible because of obviously different interventions. To minimize bias, the data analysts and personnel assessing the outcome indicators will be blinded to the group allocation during the analysis of the outcomes.
InterventionsAll participants will have their diet and exercise habits evaluated and will undergo general physical examination. A recommended caloric intake will be calculated for each participant based on their weight and self-reported physical activity levels. All participants will receive general diet and exercise suggestions according to the Dietary Guidelines for Chinese Residents. On this basis, participants in four groups will receive corresponding interventions in accordance with the research protocol.
Control groupNo additional interventions will be carried out beyond the basic health education on diet and exercise.
Structured exercise groupParticipants in the structured exercise group will engage in a structured exercise regimen at the study centre. The participants will undergo exercise training three times a week for 24 weeks. Structured exercise comprises both aerobic exercise and resistance exercise, with an interval of 5 min between them.
Aerobic exercise is performed using a same model stationary bike (SH-B3100S, Shuhua Sports Co., Ltd., Jinjiang, China). During the initial stage of power cycling, the target heart rate (THR) of each participant will be calculated using the heart rate reserve (HRR) method. In the first stage (1–6 weeks), the THR will range from 30 to 50% of the HRR. In the second stage (7–12 weeks), the THR will range from 40 to 60% of the HRR. In the third stage (12–24 weeks), the THR will range from 40 to 75% of the HRR. Aerobic training is monitored by measuring heart rate through the use of a wearable device during the exercise period. If the rating of perceived exertion (RPE) is below the specified range after three consecutive exercise sessions and the resting heart rate increases by no more than five times per minute the next day, participants will be considered to have advanced.
Participants will engage in resistance exercise by using a multi-functional trainer (Meridian M9, Meridian Fitness Equipment Co., Ltd., Wuhan, China), which includes 8 movements: high pull down, sitting rowing, sitting leg lift, standing calf bend, sitting lying push, abdominal pull down, forearm bend, and arm bend and extension. Participants will be instructed to repeat each movement 8–12 times as a set, with two sets per movement and 1–2 min of rest between sets. Participants will undergo personalized strength load assessment by trained professionals to determine the appropriate resistance load and use standardized training equipment under staff guidance. In the first stage (1–6 weeks), the load intensity will be set at 50-60% of the one-repetition maximum (1RM). In the second stage (7–12 weeks), the load intensity will be set at 60-70% of the 1RM. In the third stage (12–24 weeks), the load intensity will be set at 60-80% of the 1RM. When participants are able to perform two additional repetitions in the last set of a specific movement for two consecutive training sessions, the weight will increase in the subsequent training session.
Diet intervention groupParticipants in the diet intervention group consumed 80 g of minimally processed instant oatmeal daily for 24 consecutive weeks, which contains 3.9 g of β-glucan per 100 g (QUAKER, PepsiCo Food (China) Co., Ltd., Shanghai, China). It is recommended that participants substitute oatmeal for the main course of one meal, but consuming oatmeal at any time is also permitted. Participants are required to attend the study centre monthly to obtain oats and undergo compliance evaluations.
Combined groupParticipants in this group will participate in both a structured exercise intervention (identical to the structured exercise group) and the same oat supplementation as the diet intervention group.
Screening, assessment and follow-upAfter they provide informed consent, the participants’ demographic characteristics, medical history, concomitant medication, allergy history, dietary and exercise habits, and smoking and drinking histories will be gathered through an onsite questionnaire. The Physical Activity Readiness Questionnaire (PAR-Q) will assess the suitability of exercise for each participant. Physical examinations, laboratory examinations, gut flora and gut hormone analyses, electrocardiogram, abdominal quantitative computed tomography and body composition analyses will be performed to complete the baseline assessment. In addition, hepatitis panel will be performed on all participants to exclude viral hepatitis, serum β- human chorionic gonadotropin (hCG) test will be performed on reproductive-age female participants to exclude pregnancy. The assessment schedule is shown in Table 1.
Participants who meet all the inclusion criteria and none of the exclusion criteria will be randomized. Their muscle strength will be measured. Dietary intake will be documented in a 72-hour dietary form to analyse dietary intake and caloric consumption. Research assistants will teach them how to take pictures of their food with coordinate paper as the background. After that, they will receive a corresponding intervention for 24 weeks.
At 12 and 24 weeks after the intervention begins, participants will be investigated for dietary and exercise habits and smoking and drinking history via an onsite questionnaire. Physical examinations, laboratory examinations, gut flora and gut hormone analyses, abdominal quantitative computed tomography and body composition data will be reassessed to evaluate the efficacy of the treatments. Reproductive-age female participants will also undergo serum β-hCG tests to detect pregnancy. Adherence to the prescribed exercise and dietary regimens will be assessed, along with the safety of the intervention.
Table 1 Schedule of assessmentsAssessment of primary and secondary outcome variablesThe primary study outcome measure will be the difference in the reduction in the intrahepatic lipid content between the groups, which will be evaluated based on the results of quantitative computed tomography (QCT). A routine CT scan of the abdomen will be performed with a Canon 320-row wide-detector CT (Aquilion ONE Vision, Canon Medical Systems, Otawara, Japan). The CT scan will acquire spiral scan data (120 kV, 120–400 mA) from the lung base to the pubic symphysis in the supine position. The image will be reconstructed to 5-mm-thick and 1-mm-thick slices by the adaptive iterative dose reduction three-dimensional (AIDR3D) algorithm. Then, the images will be transferred to a workstation with a PRO Bone Mineral Densitometry System (version 5.1, 2022 Mindways Software, Inc.) to analyse the intrahepatic lipid content at baseline and after 12 and 24 weeks (Table 1).
Secondary outcomes are: (1) anthropometry, including weight, and the waist-to-hip ratio. (2) blood pressure. (3) body composition, including fat mass, lean mass and bone mass. (4) muscle strength, including grip strength, back strength, and 30-s seat test. (5) venous blood laboratory indicators, including fasting blood sugar, glycosylated haemoglobin, insulin, blood lipids, transaminase, renal function, and the bilirubin.
The exploratory outcomes include gut flora and gut hormones, such as glucagon-like peptide-1, cholecystokinin, peptide tyrosine tyrosine, and ghrelin.
Fasting blood samples will be drawn from participants in the morning after an overnight fast. The detection methods for the outcomes are shown in Table 2. Body composition will be measured using a Hologic dual-energy X-ray bone density instrument (Discovery Wi, USA). Additionally, shotgun metagenomic sequencing will be applied to faecal samples to analyse the gut flora. Gut hormones will be measured using an enzyme-linked immunosorbent assay combined with the results of the gut flora to explore possible treatment mechanisms.
Safety assessments will involve monitoring the incidence of all adverse events (AEs) and serious AEs. These events will be coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 19.1. Participants can self-report AEs at any time, and the investigator will also acquire information during study visits.
The primary, secondary and exploratory outcome variables and safety will be assessed at baseline and at 12 and 24 weeks.
Table 2 Primary and secondary outcomesMonitoring complianceDuring the exercise period, adherence assessments will be conducted weekly to track the exercise status of each participant. Participants allocated to the diet intervention group will be required to share photos demonstrating their oat consumption with researchers daily via WeChat. Compliance with the diet plan will be monitored through the completion of a 72-hour dietary record at baseline and at 12 and 24 weeks. The images of the 72-hour dietary records will be analysed by nutrition experts using Da Ying Jia Nutritionist Web Portal service (https://www.dyhomedr.com/) to assess food ingredients and calculate caloric intake.
Strategies to encourage complianceCompliance with the structured exercise and oat supplementation interventions is a considerable source of bias. During the intervention, regular telephone follow-ups will be conducted to encourage participants to engage in exercise and consume oats as described by the intervention protocol. During these phone calls, participants can communicate with researchers regarding any questions relating to the intervention and will receive advice and counselling to improve their compliance. Additionally, our exercise intervention will be conducted at a research centre that is equipped with standardized sports equipment, and professionals will be available for guidance to facilitate exercise adherence. In general, study personnel will be accessible to participants via WeChat at all times to address any concerns related to the intervention.
Sample size calculationThe sample size for this study was calculated using NCSS-PASS11 software. Based on previous research data, assuming that the mean and standard deviation of the intrahepatic lipid content changes in the structured exercise group were 7% and 5%, those in the diet intervention group were 6% and 5%, and those in the control group were 4% and 3%, respectively, at least 72 participants would be needed to provide 90% power with a two-tailed 0.05 significance level when using the two-sample t test and to maintain a ratio of 1:1 in the diet intervention group (diet intervention group + combined group) and nondiet intervention group (structured exercise group + control group). Similarly, at least 33 participants would be required in both the structured exercise group (structured exercise group + combined group) and the nonstructured exercise group (diet intervention group + control group). To maintain a ratio of 1:1:1:1 in the four groups, the maximum sample size was used, with a minimum of 36 participants needed in each group. Assuming an attrition rate of 20% over 6 months, 45 participants will be needed in each group, with a total of 180 participants in four groups.
Statistical analysisAll the statistical analyses will be conducted using SAS statistical software (version 9.3). All the statistical tests and confidence intervals will be calculated using a two-sided test with a significance level of 5%. Descriptive statistics will be used to summarize the demographic and clinical characteristics of participants randomized to the four groups. The change from baseline will be defined as the difference between the endpoint value at the specified time point and the initial baseline value.
The primary efficacy analysis for the study endpoints will be conducted via a full analysis set (FAS) and a per protocol set (PPS). The FAS will consist of all randomly assigned participants with one follow-up, which is as close as possible to the intention-to-treat approach. The PPS will consist of participants who meet the inclusion criteria, who do not meet the exclusion criteria, and who successfully complete the intervention plan. Safety assessments will comprise all randomized participants who received at least one intervention, and will involve analysing the safety endpoints.
The primary analysis will be conducted using both the FAS and PPS approaches. The analysis of the primary indicators will include a t test or Wilcoxon rank sum test to compare the changes in the intrahepatic lipid content between the structured exercise group and the nonstructured exercise group or between the diet intervention group and the nondiet intervention group; repeated measures analysis of variance was used to compare the changes in the intrahepatic fat content among the four groups. If an overall difference was found to be statistically significant, pairwise comparisons were conducted.
The analysis of the secondary indicators will include a t-test or Wilcoxon rank sum test to compare the changes in metabolic indicators between the structured exercise group and the nonstructured exercise group or between the diet intervention group and the nondiet intervention group; a mixed effects model was used to correct the influence of metabolic indicators on changes in the intrahepatic content of lipids and explore the mechanism of structured exercise and diet intervention.
The safety analysis will include adverse events of vital signs and abnormal laboratory examination results, which will be summarized in a list, and the proportion of adverse events and withdrawal due to adverse events among the four groups will be compared using a chi-square test or Fisher exact probability test.
Data managementThe data will be managed at the Beijing Friendship Hospital Pinggu Campus. Double data entry by two individuals will be conducted using an electronic data capture system. All participants will be identified by a unique number and the first letter of their name. All personal information of the participants will be kept confidential, and the cabinet where the study data are stored will be locked. In addition to the research team members, only the data monitoring committee of Peking University Clinical Research Institute and the Ethics Committee will be allowed to access the data.
Comments (0)