
Remember me
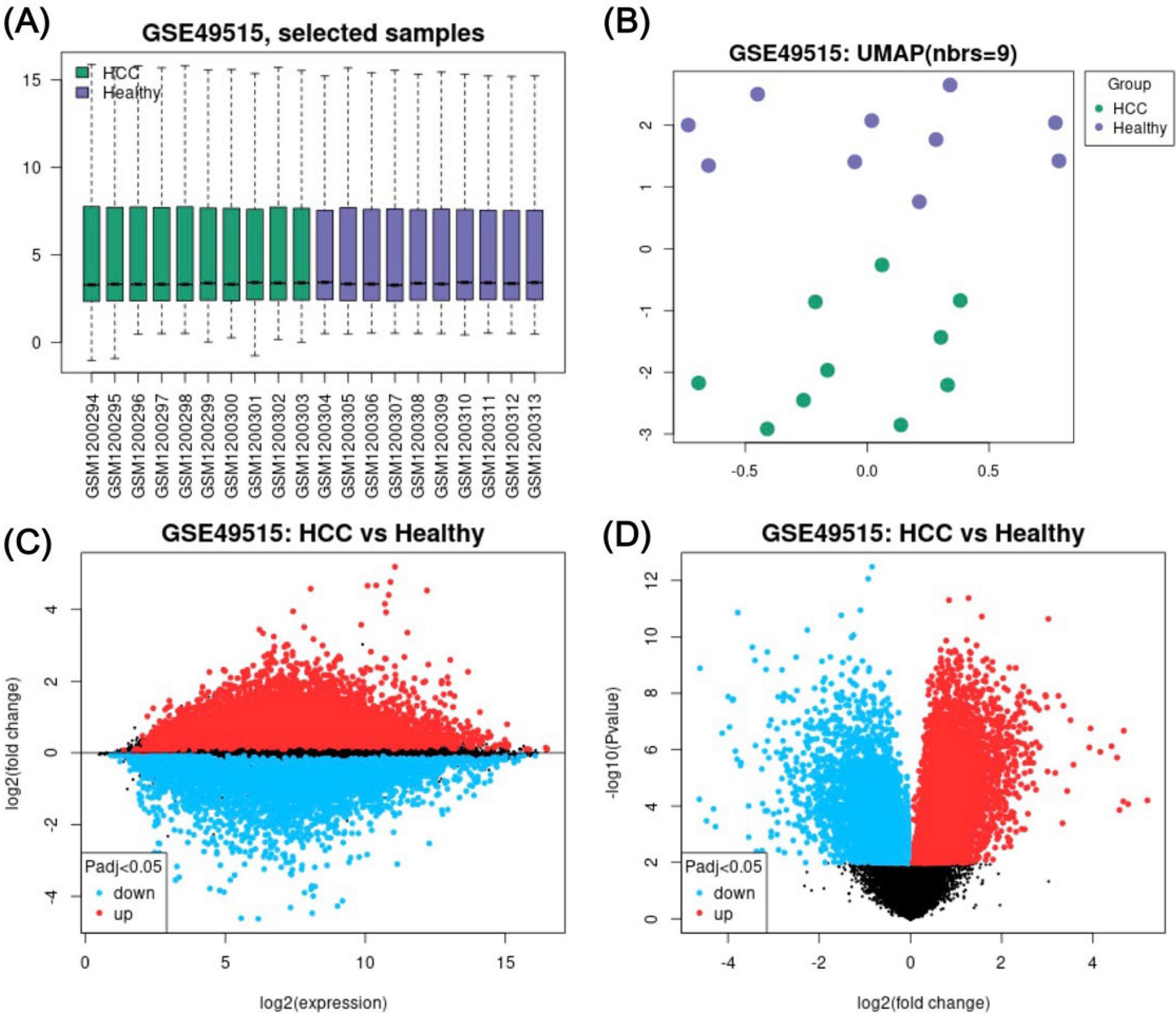






Differential gene expression (DEG) analysis revealed 10,009 DEGs between 10 HCC versus 10 control samples (Fig 1A-D). In total, 2,109 significant DEGs were determined (p-adjusted value < 0.05 and |logFC| > 1.0), consisting of 807 downregulated and 1,302 upregulated DEGs. CXCL8, HBEGF, GAS2L1, G0S2, JUN, BRE-AS1, ELF2, DAO, PHACTR1, and NR4A2 are among the top 10 downregulated DEGs discovered in HCC. In contrast, KLRD1, ITGA10, HBA2, SH3YL1, CREB5, ABAT, CCNE2, HBB, WBP2, and C8B are the top 10 upregulated DEGs.
Fig. 1
Differential gene expression analysis of GSE49515. A A Boxplot of selected samples from 10 non-HCC subjects and 10 subjects diagnosed with HCC showed the median-centred values of normalised data. B Uniform Manifold Approximation and Projection (UMAP) visualises the samples within each group. C A mean difference (MD) plot demonstrates logFC against the mean log2 expression values of the DEGs. D A volcano plot visualises the relationship between -log10 p value and logFC, representing the statistical significance and magnitude of change for DEGs. Red and blue dots signify overexpressed and underexpressed DEGs, respectively.
GO and pathway enrichment of DEGsPathway enrichment analysis revealed significant overrepresentations of BP, MF, CC pathways among the upregulated DEGs, including the top five BP: tRNA processing (GO:0008033), positive regulation of interleukin-8 production (GO:0032757), tRNA metabolic process (GO:0006399), ncRNA transcription (GO:0098781), and RNA modification (GO:0009451) (Fig. 2A); the top five MF: nuclear hormone receptor binding (GO:0035257), catalytic activity acting on RNA (GO:0140098), tRNA methyltransferase activity (GO:0008175), death receptor binding (GO:0005123), and tumour necrosis factor receptor superfamily binding (GO:0032813) (Fig. 2B); and the top five CC: late endosome membrane (GO:0031902), Cul4-RING E3 ubiquitin ligase complex (GO:0080008), endocytic vesicle (GO:0030139), early endosome (GO:0005769), and cytoplasmic vesicle lumen (GO:0060205) (Fig. 2C).
Fig. 2
GO and KEGG pathway enrichment of overexpressed DEGs. Selection of the top 10 terms was guided by a p value below 0.05. For each term, gene count signifies the total number of related genes.
In addition, the downregulated DEGs are also involved in several BPs, such as cell cycle arrest (GO:0007050), regulation of RNA splicing (GO:0043484), regulation of mRNA splicing via spliceosome (GO:0048024), positive regulation of establishment of protein localisation (GO:1904951), and positive regulation of cellular protein localisation (GO:1903829) (Fig. 3A). The MF and CC of the downregulated DEGs were also predominantly enriched in several molecular pathways and cellular constituents, such as the DNA-binding transcription factor binding (GO:0140297), RNA polymerase II-specific DNA-binding transcription factor binding (GO:0061629), glucocorticoid receptor binding (GO:0035259), nuclear hormone receptor binding (GO:0035257), hormone receptor binding (GO:0051427), nuclear speck (GO:0016607), transcription regulator complex (GO:0005667), RNA polymerase II transcription regulator complex (GO:0090575), PML body (GO:0016605), and beta-catenin/TCF complex (GO:1990907) (Fig. 3B and C). Interestingly, several pathways associated with cancer were also enriched. For example, the NOD-like receptor signalling pathway, Toll-like receptor signalling pathway, FoxO signalling pathway, PDL1 expression and PD-1 checkpoint pathway in cancer, as well as a number of malignancies, for instance, bladder carcinoma, renal cell carcinoma, and colorectal cancer (Fig. 2D and 3D). Taken together, the results indicate that the DEGs could activate several important processes and pathways linked to HCC.
Fig. 3
GO and KEGG pathway analysis of underexpressed DEGs. Identification of the 10 highest-ranking terms was guided by a p value below 0.05. For each term, gene count signifies the total number of related genes.
DEG-based PPI network and cluster evaluationThe physical interactions among the protein-encoded genes of HCC were retrieved using StringApp. In total, 1,591 out of 1,719 DEGs were constructed into the DEG-based PPIN. Seven hundred and seven (707) single nodes (DEGs) were removed, leaving the PPIN with 884 nodes and 2,486 edges (interactions). We subsequently implemented the MCODE approach to identify the densely interacted domains of the PPIN, resulting in 42 clusters. However, we specifically chose the top five clusters with the strongest scores and highest number of DEGs for subsequent analysis. A total of 87 core genes were identified, with cluster 5 containing the most genes (n=38), followed by cluster 1 (n=21), clusters 2 (n=10), clusters 3 and 4 (n=9) (Table 1).
Table 1 Top five clusters identified using MCODE.Prognostic association of core gene expressions with survival outcomesThe survival patterns of HCC cases documented in the TCGA database were analysed based on the identified core genes using GEPIA2. The findings indicated that the 25 core gene overexpressions were linked to statistically significant shorter OS. In our study, 10 upregulated core genes from cluster 1 were significantly implicated in diminished overall survival probability, including GTF2H1 (HR=1.8, p=0.011), MED17 (HR=1.6, p=0.011), HIST1H4H (HR=1.8, p=0.030), MED8 (HR=2, p<0.001), PLRG1 (HR=1.4, p=0.041), POLR2H (HR=1.8, p=0.002, ), SF3A3 (HR=1.9, p<0.001), SF3B3 (p=0.011, HR=1.6), TAF13 (HR=1.5, p=0.025), and CRNKL1 (HR=1.9, p<0.001) (Fig. 4). High DCLRE1C (HR=1.5, p=0.020), XRCC5 (HR=1.5, p=0.016), and FANCI (HR=1.6, p=0.007) expressions in cluster 2 were also associated with worse survival (Fig. 5). Out of nine core genes in cluster 4, MRPL33 (HR=1.6, p=0.005), MRPL35 (HR=1.5, p<0.001), MRPL50 (HR=1.5, p=0.030), and MRPS30 (HR=1.7, p=0.003) exhibited significant relationships with unfavourable HCC prognosis. Furthermore, seven cluster 5 core genes, named ATG7 (HR=1.6, p=0.014), DCAF7 (HR=1.6, p=0.009), SRSF10 (HR=1.6, p=0.008), FXR1 (HR=1.5, p=0.024), DCAF16 (HR=1.7, p=0.004), DCAF13 (HR=1.5, p=0.016), and DCAF10 (HR=1.5, p=0.037), were linked to adverse OS in HCC cases. In summary, the established 25 core genetic elements might be potential minimally invasive prognosticators for HCC.
Fig. 4
The prognostic properties of core genes for cluster 1 on overall survival of HCC patients using the GEPIA2 platform. Kaplan–Meier survival curves for GTF2H1, MED17, HIST1H4H, MED8, PLRG1, POLR2H, SF3A3, SF3B3, TAF13, and CRNKL1 in HCC patients were constructed based on the TCGA data. Red nodes denote overexpressed genes, and the blue nodes denote underexpressed genes.
Fig. 5
The prognostic core genes for clusters 2, 4, and 5 in liver HCC using the GEPIA2 platform. The Kaplan–Meier survival curve for DCLRE1C, XRCC5, and FANCI (cluster 2); MRPL33, MRPL35, MRPL50, and MRPS30 (cluster 4); and ATG7, DCAF7, SRSF10, FXR1, DCAF16, DCAF13, and DCAF10 (cluster 5) in patients with HCC from TCGA. The red node represents an overexpressed gene, and the blue node represents an underexpressed gene.
The analysis of immune cell trafficking based on core genesThe (i) correlation and (ii) relationship between the mRNA expression and somatic copy number alteration (SCNA) of 25 core genes and trafficked immune cells within the HCC microenvironment were analysed using TIMER. SCNA compares tumour infiltration levels in HCC based on different SCNAs for 25 core genes. Also, genes with weak correlations (ranging from 0.2 to 0.4) were subsequently removed, leaving only those displaying significantly considerable mRNA and CNA correlations with infiltrating immune cells in HCC as critical candidates. Therefore, six core genes were identified as strong candidates for non-invasive prognostic biomarkers: GTF2H1, SF3A3, FANCI, XRCC5, DCAF7, and DCAF16. GTF2H1 demonstrated moderate correlations with macrophage (Spearman’s ρ = 0.533, p <0.001) and neutrophil abundance (Spearman’s ρ = 0.467, p <0.001) (Fig. 6A). SF3A3 exhibited a moderate correlation with macrophage (Spearman’s ρ = 0.443, p<0.001), neutrophils (Spearman’s ρ = 0.422, p<0.001), and dendritic cells (Spearman’s ρ = 0.473, p<0.001) trafficking (Fig. 6B), whereas FANCI with B Cell (Spearman’s ρ = 0.489, p <0.001), macrophage (Spearman’s ρ = 0.491, p <0.001), neutrophils (Spearman’s ρ = 0.416, p < 0.001), and dendritic cells (Spearman’s ρ = 0.489, p<0.001) infiltration (Fig. 6C). XRCC5 also moderately correlated with the macrophage (Spearman’s ρ = 0.452, p<0.001), neutrophils (Spearman’s ρ = 0.454, p 0.001), and dendritic cells (Spearman’s ρ = 0.458, p<0.001) (Fig. 6D). Two DCAF genes, DCAF7 and DCAF16, demonstrated a moderate correlation with various immune cells: DCAF7 with macrophages (Spearman’s ρ = 0.453, p <0.001), neutrophils (Spearman’s ρ = 0.429, p<0.001), and dendritic cells (Spearman’s ρ = 0.429, p <0.001), and DCAF16 with CD4+ T cells (Spearman’s ρ = 0.452, p<0.001), macrophages (Spearman’s ρ = 0.519, p <0.001), neutrophils (Spearman’s ρ = 0.475, p<0.001), and dendritic cells (Spearman’s ρ = 0.438, p <0.001) (Figs. 6E-6F). These findings offer compelling support that GTF2H1, SF3A3, FANCI, XRCC5, DCAF7, and DCAF16 play significant roles in immune cell trafficking, particularly macrophages, neutrophils, and dendritic cells.
Fig. 6
Correlation analyses between mRNA expression levels of A GTF2H1, B SF3A3, C FANCI, D XRCC5, E DCAF7, and F DCAF16 and the levels of immune cell trafficking within HCC tissues.
For GTF2H1, the arm-level gain of CNA is significantly related to the extent of B cell trafficking (Fig. 7A). Furthermore, the CNA (arm-level deletion) of SF3A3 was also associated with infiltrating both CD8+ T cells and neutrophil infiltration of the tumour microenvironment (Fig. 7B). Arm-level deletion of FANCI was also discovered to result in different degrees of B cells and dendritic cell infiltration (Fig. 7C), whereas arm-level deletion of XRCC5 influenced dendritic cell infiltration (Fig. 7F). In addition, high amplification of DCAF16 resulted in significantly enhanced CD8+ T cells, CD4+ T cells, and macrophage trafficking within the HCC microenvironment (Fig 7D). High amplification of DCAF7 was also associated with enhanced levels of infiltration by the CD4+ T cells, macrophages, neutrophils, and dendritic cells, whereas arm-level gain of DCAF7 led to enhanced CD8+ T cells infiltration (Fig. 7E).
Fig. 7
The relationship between somatic copy number variations (SCNA) and A GTF2H1, B SF3A3, C FANCI, D DCAF16, E DCAF7, and F XRCC5 with immune cell trafficking in HCC. The SCNA of core genes was across five different tiers: deep deletion, arm-level deletion, normal, arm-level gain, and high amplification.
We then conducted correlation analyses between GTF2H1, SF3A3, FANCI, DCAF16, DCAF7, and XRCC5 and 45 genetic markers across various immune cell populations within HCC (Table 2). Here, we found that GTF2H1 was moderately correlated with the M1 macrophage marker IRF5 (Spearman’s ρ = 0.462), dendritic cell marker NRP1 (Spearman’s ρ = 0.486), and Type 1 T helper maker STAT1 (Spearman’s ρ = 0.477). Besides GTF2H1, FANCI, and DCAF16 were also moderately correlated with IRF5, with correlation values of 0.429 and 0.431, respectively. Meanwhile, SF3A3 correlated with NRP1 (Spearman’s ρ = 0.415). The PDL1 marker CD274 was found to correlate with SF3A3 (Spearman’s ρ = 0.407), DCAF7 (Spearman’s r = 0.407), and XRCC5 (Spearman’s ρ = 0.402) in HCC. In addition, STAT1 was significantly correlated with DCAF16 (Spearman’s ρ = 0.474), DCAF7 (Spearman’s ρ = 0.422), and XRCC5 (Spearman’s ρ = 0.488). The GTF2H1 expression levels exhibited significant correlations with M1 macrophages, dendritic cells, and Th1 cell markers; SF3A3 with dendritic cells and PDL1; FANCI with M1 macrophages; DCAF16 with M1 macrophages, Th1 cells, Th2 cells, and Tregs; and DCAF7 and XRCC5 with PDL1 and Th1 cells.
Table 2 Correlation analysis between GTF2H1, SF3A3, FANCI, DCAF16, DCAF7, and XRCC5 with related immune markers in immune cells. * represents statistically significant correlationImmunohistochemical expression of core genesUsing the Human Protein Atlas (HPA) protein expression data, immunohistochemistry findings confirmed that GTF2H1, SF3A3, FANCI, and XRCC5 exhibited increased expressed protein levels in HCC compared to healthy hepatocytes (Fig. 8). When combined with the earlier analytical findings from the PBMC dataset, these results provide robust evidence that GTF2H1, SF3A3, FANCI, and XRCC5 are not only differentially expressed but also hold significant potential as minimally invasive prognosticators in hepatocarcinoma, reinforcing their relevance across both tissue-based and peripheral blood-based analyses.
Fig. 8
Immunohistochemistry (IHC) of six core genes. Protein levels of six core genes (A) GTF2H1, (B) SF3A3, (C) FANCI, (D) XRCC5, (E) DCAF7, and (F) DCAF16 were determined in (i) normal hepatic tissue and (ii) HCC tissue.
Comments (0)