
Remember me
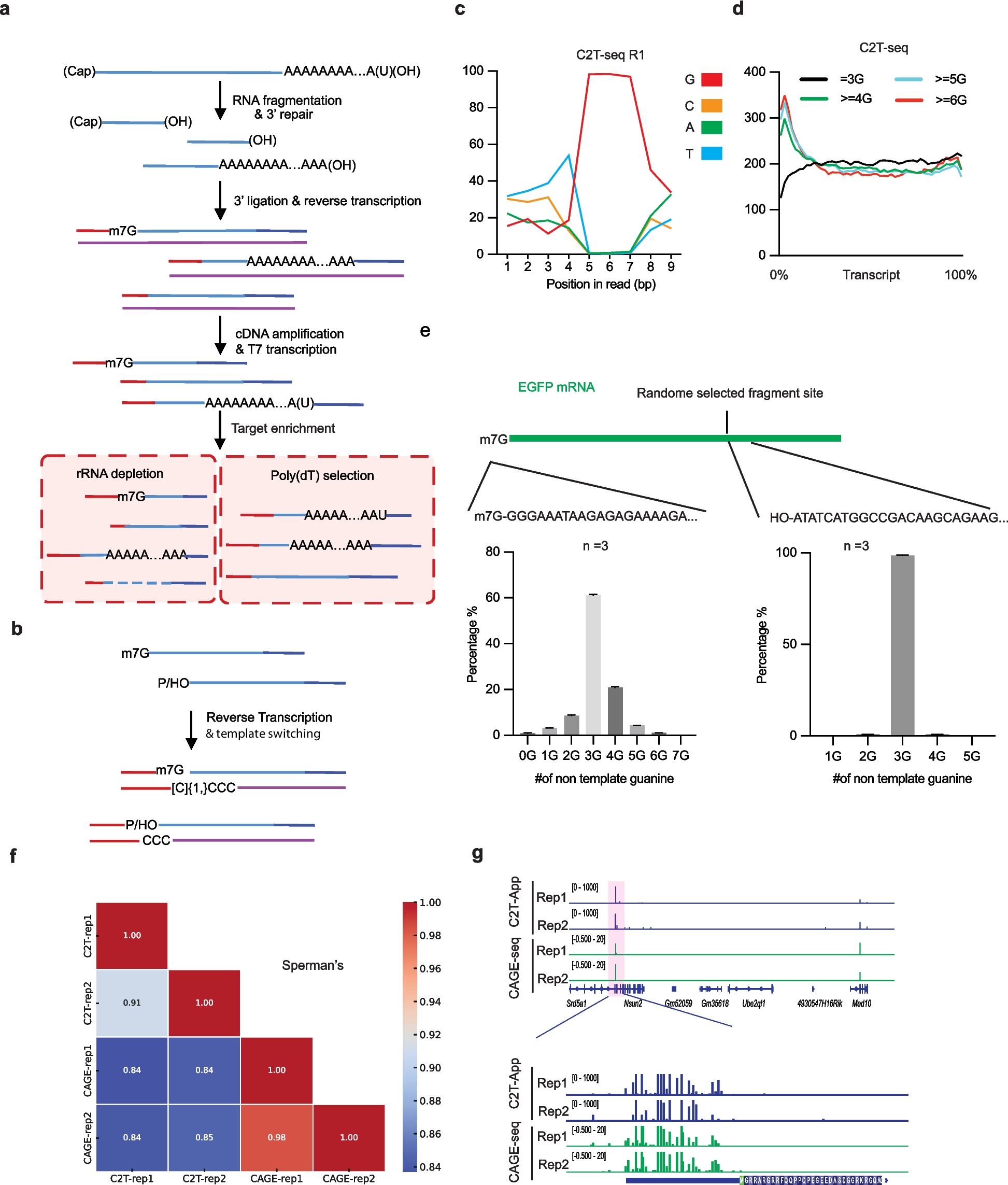






annATAC is a deep learning method based on language model (Bidirectional Encoder Representations from Transformers, BERT) and is applied to the annotation task of scATAC-seq data. The model mainly consists of five stages (Fig. 1). In stage 1, comprehensive data collection and peak island-based pre-processing. We collected scATAC-seq data covering 13 types of human tissues from the GEO database, including cancer samples and normal samples. On the premise of maximizing the preservation of the original open information of the data, these data were processed into the form of the cell-peak island to serve as the input data for the model (for detailed processing strategies, please refer to “ Methods”). In stage 2, data masking. Specifically, this study divided the expression values of peak islands into five categories and then randomly masked them. During the masking process, the positions with an expression value of 0 were ignored. In stage 3, unsupervised pre-training, which was carried out with a large amount of unlabeled scATAC-seq data. annATAC takes the BERT as its basic architecture, but due to the characteristics of scATAC-seq data, it uses the multi-head attention mechanism in Linformer [45] instead of the original BERT. Through the learning of masked positions, the model can effectively learn the interaction relationships between peak islands during the pre-training stage, which plays a crucial role in the downstream cell type annotation. In stage 4, supervised fine-tuning. After the pre-training is completed, the model has already gained a relatively in-depth understanding of the interactions between peak islands. In this stage, a small amount of labeled data is used to conduct secondary training on the model, prompting the model to be further optimized so that it can accurately identify cell types and finally complete the construction of the model. In stage 5, biological analysis. The trained model is used to conduct multiple biological analyses, such as predicting novel cell types. For the specific design of the experiment, please refer to the “ Methods” section for details.
Fig. 1
Overview of the annATAC. In step 1, collect millions of scATAC-seq data, covering 13 kinds of tumor and normal tissues in the Homo sapiens. Then further convert these data into the form of a cell-peak island for the input of the model. In step 2, perform data masking and ignore the positions where the expression values are zero. In step 3, conduct unsupervised pre-training. Build an Encoder by introducing linear transformation into the multi-head attention mechanism. Train the model by minimizing the loss between the model’s output and the ground truth before masking. In step 4, carry out supervised fine-tuning. Through secondary training on a small amount of labeled data, the model is fully converged. In step 5, conduct downstream biological analysis
Performance evaluation of cell type annotationComparative analysis with baseline methodsTo evaluate the performance of annATAC in annotating cell types, this study conducted performance evaluations on eight tissues of human adults (Breast, Stomach, Esophagus Muscularis, Lung, Transverse Colon, Gastrocnemius Muscle, Heart Right Atrial Appendage, and Heart Left Ventricle). As shown in Fig. 2A, the Accuracy (ACC), Jaccard-weighted, and Cohen’s kappa of annATAC are superior to the comparison methods in the vast majority of tissues and comparable to Cellcano in some tissues. Specifically, on the Esophagus Muscularis data with the largest number of cells, annATAC had a significant advantage in ACC, while on the Stomach data with the smallest number of samples, its ACC was slightly lower than that of Cellcano. We believe that this phenomenon is caused by the high sparsity of small sample data. The evaluation results of the Heart Right Atrial Appendage and Heart Left Ventricle tissues are shown in Additional file 1: Fig. S1A. Compared with Cellcano, annATAC had a higher ACC in predicting T cell and Smooth Muscle cells with smaller sample sizes. To comprehensively display the comparison results with the baseline methods, this study plotted the overall distribution map of ACC on the eight tissues in Fig. 2B. In summary, annATAC’s ACC performance is better that of the baseline method, with Cellcano ranking second, followed by RAINBOW. The overall distribution of Jaccard (weight) and F1 score is shown in Additional file 1: Fig. S1B.
Fig. 2
Performance evaluation of annATAC in annotating cell types. A Evaluation of the annotation performance of ACC, Jaccard_weighted, and Jaccard_micro on the scATAC-seq data of six adult tissues. The abscissa indicates the number of cells in each tissue. B Overall distribution performance evaluation of ACC on the scATAC-seq data of eight adult tissues. C t-SNE visualization display on the Heart and Stomach Tissues, where the first column shows the annotation of the original class labels. D Evaluation of annATAC’s performance in detecting novel cell types. The red color in the Sankey diagram represents novel cell types, with the real cell labels on the left side and the labels predicted by annATAC on the right side
In addition, we visualized the annotation results on the Heart and Stomach tissues (Fig. 2C). The first column of this figure shows the cell labels provided by the original study. Through experimental comparison, it was found that only annATAC and Cellcano could better restore the annotations of cell labels, and annATAC performed even better. It is particularly worth noting that annATAC performed excellently in predicting cell labels with a small number of cells. For example, in the Heart tissue, annATAC accurately predicted T cells. Identifying T cells is of crucial importance for gaining a deeper understanding of the immune microenvironment of cardiac tissue as well as the mechanisms of disease occurrence and development. For the visualization of the annotation results of other tissues, please refer to Additional file 1: Fig. S1 C.
annATAC can identify novel cell typesannATAC has a built-in option for identifying novel cell types. When the confidence level of a predicted cell is lower than the default confidence score, the model will label this cell as a novel cell type. This study tested the ability of annATAC to explore novel cell types in Adipose Omentum tissue. Compared to the previously tested and compared tissues, “adipocyte” in this tissue is a novel cell type. The experimental results are shown in Fig. 2D. annATAC can accurately predict common cell types. For the novel cell type Adipocyte, the model predicted most of these cells as “Novel_cell_type,” but a small number were predicted as Fibroblasts. Considering that both Fibroblasts and Adipocytes originate from Mesenchymal Stem cells and there are many overlaps in their differentiation regulatory mechanisms, we speculate that this is the reason why annATAC predicts some Adipocytes as Fibroblasts. Through this experiment and combined with prior biological knowledge, it can be demonstrated that annATAC can identify novel cell types.
Performance evaluation of cell subtype annotationThe differences in chromatin accessibility among different cell subtypes are relatively subtle. Therefore, compared with the annotation of rough cell types, the annotation of cell subtypes is more difficult. This study conducted experiments on different Neuronal cell subtypes of patients with Alzheimer's Disease (AD) (Astrocytes (ASC), Excitatory Neurons (EX), Olig.progenitors (OPC), Inhibitory Neurons (INH), Microglia (MG), Oligodendrocytes (ODC), and Pericytes/Endothelial (PER.END)). We collected a total of 9 datasets from the GEO database [46], including 7 datasets of AD patients and 2 datasets of the control group.
annATAC accurately annotates cell types across different data scalesAmong the nine collected datasets, we selected two datasets with the smallest (2558) and the largest (9761) number of cells respectively to evaluate the annotation results. As shown in the radar chart in Fig. 3A, after comprehensively evaluating with five metrics, it was found that regardless of the data scale, annATAC always maintained a relatively high annotation ACC, and Cellcano still ranked second in terms of performance. Secondly, to further and comprehensively observe the comparison results between annATAC and other algorithms, we used semi-violin plots to display the ACC and F1 score of annATAC and other algorithms (Fig. 3B). It can be seen from the figure that annATAC had an excellent performance in ACC, and its F1 score could reach 90%. It can thus be concluded that annATAC had a relatively high ACC in predicting cell subtypes. In addition, Additional file 1: Fig. S2 presents the experimental results of another three metrics, namely Cohen’s kappa, Jaccard_weighted, and Jaccard_micro. Through comparison, the superiority of annATAC could also be confirmed.
Fig. 3
Performance evaluation of cell subtype annotation. A Comparing annATAC with other comparative algorithms in the AD datasets with the smallest sample size (2588) and the largest sample size (9761). B Semi-violin plots display the performance of annATAC and other algorithms in terms of ACC and F1 score on seven AD datasets with different sample sizes. C The experimental performance of two AD and control groups is evaluated in terms of ACC and F1 score. D A comparison displays the annotation results of annATAC and those of RAINBOW and Cellcano on four AD and one control datasets. The bottom row indicates different tissues and the number of cells. E Performing t-SNE visualization on the AD datasets shows that different colors represent different cell types
annATAC has superiority in identifying cell types in Alzheimer’s diseaseThis study collected two sets of control experiment data, which were respectively from 2 AD patients and two healthy control groups. The ages of the patients were all around 80 years old. The same sequencing technique (Illumina NovaSeq 6000) was adopted for both groups, and the sequencing sites were all in the Frontal Cortex region. The experimental results are shown in Fig. 3C. Through a comparative analysis using ACC and F1 score, we found that annATAC had a significant advantage in ACC when predicting the cell types of AD patients and was nearly 10 percentage points higher than Cellcano in ACC. While in the control group, the ACC of Cellcano was only around 5 percentage points lower than that of annATAC.
Comparative evaluation with other methodsWe presented the prediction results of different algorithms on AD datasets of 5 patients and one control group dataset. Among them, Cellcano and RAINBOW algorithms with better performance were selected for comparison with our method. As shown in Fig. 3D, annATAC demonstrated superiority in most tissues. However, on the control group data, its performance was slightly lower than that of Cellcano. In addition, we selected the prediction results of patient 2 with AD for visual display, as shown in Fig. 3E compared to Cellcano, annATAC had a higher ACC in annotating cell labels. Cellcano mispredicted some ODCs as EXs. Moreover, in the t-SNE plot, it could be observed that Cellcano failed to separate well the regions where cell types were interconnected, while annATAC performed better in this regard.
Biological analysis of Alzheimer’s diseaseTo deeply explore the analytical potential of annATAC, we carried out a series of biological analyses on patient 4 with AD. Through multi-dimensional experimental investigations and precise analyses, it is strongly demonstrated that annATAC has significant potential in identifying marker peaks and marker motifs, and is expected to provide new technical support and research perspectives for relevant biological research and disease analysis.
annATAC has the potential to identify marker peaksThis study focused on the biological analysis of patient 4 with AD. Firstly, the cell type annotation results were presented using Uniform Manifold Approximation and Projection (UMAP), as shown in Fig. 4A on the left side of the figure, the FindClusters function in the Seurat tool [26] was used for cell clustering. On the right side, the prediction results of annATAC were labeled for visual presentation. Through the UMAP visualization analysis, it was observed that annATAC had very distinct boundaries for the annotation and classification of cell types. In particular, it was able to accurately annotate the easily confused Neuron types EX and INH. In addition, Fig. 4B shows the proportion of regulatory elements in different cell types, with UnionPeaks presenting the proportion of overlapping regulatory elements. Subsequently, the getMarkerFeatures function in the ArchR tool [27] was used to identify the specific marker peaks in different cell types (Fig. 4C). From the experimental results, it could be seen that the peak enriched in the LHX families was the marker peaks of ASC. As shown in the graphs on the left side of Fig. 4C, the corresponding marker peak location information could be found at the corresponding positions on chromosome 1. Meanwhile, it was found that the peak enriched in the NEUROG families was the marker peaks of EX, which were presented in the graphs on the right side of Fig. 4C. Our findings are consistent with existing research results [47, 48]. In addition, the other peak enriched by the ARID family is the marker peak of ASC, and the other peak enriched by the TCF family is the marker peak of EX (Additional file 1: Fig. S3 A). The peak enriched in the SOX family were the marker peak of ODC, and the peak enriched in CD68 were the marker peak of MG. These findings not only deepened our understanding of the characteristics of cell types in AD patients, but more importantly, provided crucial technical support for the targeted treatment of AD, and were expected to promote the further development of the AD treatment field.
Fig. 4
Biological analysis of AD. A UMAP visualization of annATAC’s cell type annotation for patient 4 with AD. B Proportions of regulatory elements contained in different cell types. C Display of marker peaks on different cell types, with red auxiliary lines indicating the positions of marker peaks. D Display of marker motifs on different cell types
annATAC has the potential to identify marker motifsTo further explore the regulatory mechanisms specific to different cell types, we carried out a marker motif analysis (Fig. 4D). During the research process, by combining the analysis results with Fig. 4A, we confirmed that LHX9, NEUROG3, SOX13, TCF7L1, and KLF2 are the marker motifs of ASC, EX, ODC, PER.END, and MG respectively. Furthermore, to more accurately depict the specificity of the marker motifs, we used mountain plots for visual display. For specific details, please refer to Additional file 1: Fig. S3B. Through the mountain plots, the distribution characteristics and specificity of the marker motifs of different cell types can be presented intuitively, providing a clear and powerful basis for further research on cell regulatory mechanisms.
Cell type-specific analysisTo further reveal the regulatory mechanism of transcription factors, we carried out transcription factor footprint analysis using the getFootprints function in the ArchR tool, and the experimental results are presented in Fig. 5A. In the upper part of Fig. 5A, the transcription footprints of the SOX4 and SOX17 enriched in ODC are clearly shown. This presentation intuitively presents the action sites and patterns of these two transcription factors in the gene regulatory regions related to ODC cells. The lower part focuses on PER.END and presents the transcription footprints of the SMARCC family enriched in this cell type. Through the analysis above, we can accurately locate the binding positions of transcription factors in gene promoters and other key regulatory regions, especially the binding situation near the transcription start site (TSS), providing key information for analyzing the initiation mechanism of gene transcription. Meanwhile, this analysis can also deeply reveal the binding patterns of transcription factors to DNA and the interrelationships among different transcription factors. These findings are crucial for understanding the precise regulation of gene expression and provide valuable clues for the study of disease pathogenesis, which are expected to promote the in-depth development of related disease research fields. In addition, we conducted cell trajectory analysis on PER.END cells in both motif and peak dimensions (Fig. 5B). In this analysis, we used MotifMatrix and PeakMatrix to construct cell trajectories. The motif matrix contains information about transcription factor binding motifs, while the peak matrix reflects the peak characteristics of chromatin accessibility. By using these two matrices to construct cell trajectories, we can deeply analyze the impact of dynamic changes in transcription factor binding behavior on cell development trajectories during processes such as cell differentiation. Starting from the key aspect of transcriptional regulation helps us to have a more comprehensive and in-depth understanding of the dynamic changes in cells at different stages, providing important clues for revealing the molecular mechanisms of cell differentiation.
Fig. 5
A Analysis of cell type-specific transcription factor footprints in AD patients. B Pseudo-time trajectory analysis of marker motifs and marker peaks in AD patients
Model robustness testingannATAC has strong generalization abilityTo further investigate the generalization ability of the model to noise, this study conducted experiments on five tissues: Breast Mammary tissue, Transverse Colon, Esophagus Muscularis, Lung, and Stomach (Fig. 6A). Firstly, we fine-tuned the model using the original labels (corresponding to the RAW in the first column of the figure) and tested the annotation ACC on the aforementioned 5 tissues. Subsequently, to simulate noises, we randomly shuffled the fine-tuned data labels at ratios of 5%, 10%, 15%, and 20%, and then conducted training in sequence (corresponding to columns 2, 3, 4, and 5 in the figure), finally predicted the annotation ACC for each scenario. From the experimental results, it can be observed that regardless of the proportion of label shuffling or the number of cells, the ACC of the model prediction remains relatively stable, and the trend of change shows significant consistency. This phenomenon fully demonstrates that annATAC has a strong generalization ability and can effectively resist the interference of data noise. In addition, we further expanded the scope of the experiment by performing a 10% random shuffling operation on the fine-tuned labels on 8 tissues, then comparing them with the results obtained from training with the original labels. The results show that the trends of the two are consistent, and the specific experimental results are detailed in Additional file 1: Fig. S4 A.
Fig. 6
Model stability testing. A Model robustness testing was conducted on five tissues: Breast Bammary tissue (cell: 8547), Transverse Colon (cell: 6499), Esophagus Muscularis (cell: 10,783), Lung (cell: 3313), and Stomach (cell: 1266). RAW indicates that the labels were not shuffled during the fine-tuning process, dis_5 indicates that 5% of the labels were randomly shuffled, dis_10 indicates that 10% of the labels were randomly shuffled, and so on. B Randomly select 40%, 60%, and 80% of the fine-tuning data for training, and test the annotation ACC on two tissues, Gastrocnemius Muscle and Lung. C Pre-training ablation experiments. The upper part shows ACC and loss with pre-training, while the lower part shows the results without pre-training
annATAC has a relatively strong adaptabilityIn order to further test the adaptability of the model to fine-tuning data volume, this study conducted specialized adaptability tests on two organizations, the Gastrocnemius Muscle and Lung. In the experimental operation, we strictly extracted 40%, 60%, and 80% of the data from the fine-tuning data for fine-tuning of the model. After completing the fine-tuning, the model was tested on the corresponding datasets of these two organizations to verify its adaptability to different amounts of data. To ensure the reliability and stability of the experimental results, three rounds of experiments were independently repeated for each subset of data at different ratios. The experimental results in Fig. 6B comprehensively evaluate the performance of the model under different data volume adjustments using five evaluation metrics. Meanwhile, t-test statistical methods were adopted to accurately detect the differences in prediction ACC corresponding to different proportions of fine-tuned data. The clear trend of changes presented in the graph, as well as the statistically significant p values displayed, demonstrates that annATAC exhibits strong adaptive data volume capabilities. This means that the model does not overly rely on fine-tuning the amount of data when making predictions and can maintain stable and efficient performance at different scales of fine-tuning data.
Ablation experimentTo enhance the interpretability of the model, we carried out ablation experiments (Fig. 6C). In this ablation experiment, we set the training period to 15 epochs. The upper part of Fig. 6C shows the changes in ACC and loss after introducing the pre-trained model for fine-tuning, while the lower part presents the corresponding ACC and loss results when directly training on the fine-tuning dataset without introducing the pre-trained model. It can be seen that after introducing the pre-trained model, the ACC at epoch 0 is 0.67, while without introducing the pre-trained model, the ACC at epoch 0 is only 0.32. This significant difference indicates that the pre-trained model provides a good foundation for the subsequent fine-tuning. In terms of the convergence speed of the model, the fine-tuning process with the addition of the pre-trained model converges relatively quickly. To sum up, pre-training plays a crucial role in the prediction performance of the model. In addition, we also conducted a comparative analysis of the F1 score before and after the ablation (Additional file 1: Fig. S4B).
Comments (0)