
Remember me
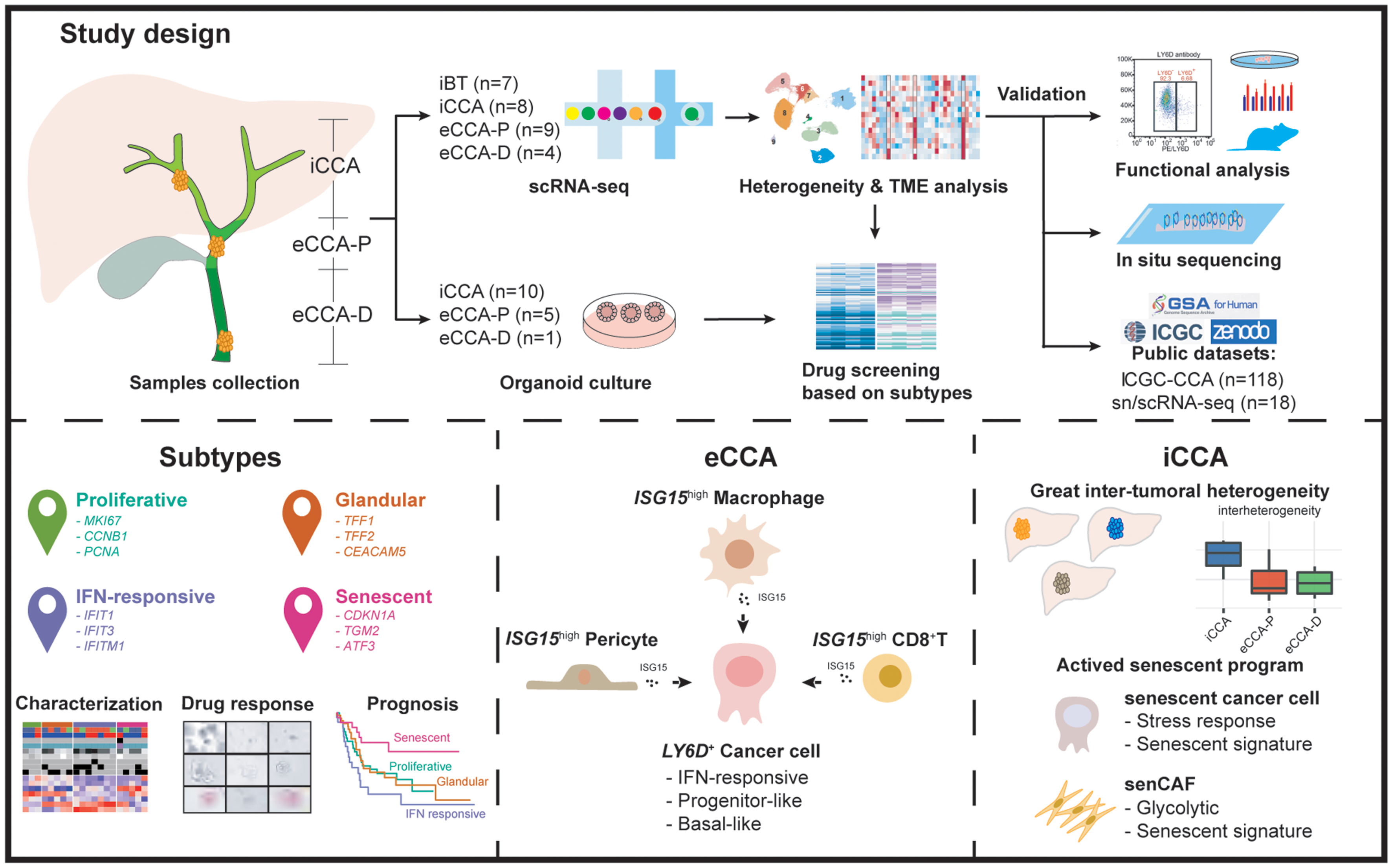


We constructed a single-cell atlas using fresh specimens from a cohort of 28 treatment-naïve patients, which spanned diverse anatomical and pathological contexts, including seven inflammatory EBTs (iBT1-7, mainly associated with choledolithiasis or choledochal cysts), eight iCCAs (iCCA1-8), nine eCCA-Ps (eCCA-P1-9), and four samples of eCCA-Ds (eCCA-D1-4) (Fig. 1A; Table S1). We profiled 109,071 cells encompassing various cell types: epithelial cells (EPCs, EPCAM+KRT19+), endothelial cells (PECAM1+VWF+), mesenchymal cells (LUM+COL1A1+), Schwann cells (S100B+GAP43+), myeloid cells (CD163+CD68+), B cells (CD79A+MS4A1+), plasma cells (CD79A+MZB1+), T/NK cells (CD3D+NKG7+), and mast cells (KIT+CPA3+) (Figs. 1B-D, and Fig. S1). These major subsets consistently appeared across multiple samples but with uneven distribution (Figs. 1E-F). Schwann and mast cells were notably enriched in extrahepatic lesions. Tumor samples showed a pronounced infiltration of lymphocytes. Myeloid cell enrichment in iCCAs likely implied a unique Kupffer cell-associated immune niche. The desmoplastic nature of eCCA-Ps was reflected by the disproportionate distribution of epithelial and mesenchymal cells [7].
Discriminating malignant from non-malignant EPCs and characterizing precancer cell StatesWe stratified EPCs (n = 20,052) into malignant and non-malignant cells using the inferCNV algorithm, identifying 20 subclones. Subclones 3, 5, and 6, predominantly from benign inflammatory bile ducts, were designated non-malignant based on low CNV scores (Figs. S2A-C). Cells with chromosomal alterations in other subclones, primarily from CCA samples, were classified as malignant EPCs, exhibiting distinct CNV profiles and salient patient occupancy (Fig. S2D).
Chronic inflammation is the hallmark of BTC oncogenesis [8], yet the precancerous epithelial states of BTCs remain poorly understood. By including samples primarily from patients with choledolithiasis or choledochal cyst, our data provides a unique opportunity to delineate the precancerous states of bile ducts, with particular relevance to BTC etiologies in East Asia [9]. We categorized non-malignant EPCs (nEPCs) into eight subtypes: gastric-like (PGC, MUC6), intestinal-like (TFF1, SPINK4), basal-like (KRT13, LY6D), mesenchymal-like (FN1, VCAN), inflammatory (CST1, CXCL1/8), immune-regulating (SREPING1, HLA-A), proliferative (CCNB2, MKI67), and developmental (FOLR1, FGFR2) (Figs. S3A, B). Pathway enrichment analysis confirmed these functional distinctions (Fig. S3C; Table S3). Gastric-like, intestinal-like, and inflammatory nEPC subtypes were more prevalent in benign inflammatory lesions, implying early hijacking of normal regenerative pathways [6]. By contrast, CNVlow nEPCs within tumors were dominated by mesenchymal-like, developmental, and immune-regulating cell states, which were preferentially enriched in iCCAs, eCCA-Ps, and eCCA-Ds, respectively (Fig. S3D). These findings implied that carcinogenesis across different CCA subtypes is likely driven by differential processes such as epithelial-mesenchymal plasticity (EMP), onco-fetal reprogramming, and dysregulated immunosurveillance. By depicting the heterogeneity of precancerous epithelial states during the inflammation-carcinogenesis cascade and across the biliary tree, our data offers insights into the biliary-specific precancer hallmarks [6].
Decoding shared and distinct malignant meta-programs across CCA subtypesNext, we focused on cancer cells to explore whether different CCA subtypes exhibit shared or subtype-specific cell states. Using the NMF method, we identified seven robust meta-programs (MPs) (Figs. 2 A and S4A; Table S4). The co-existence of mosaic MPs within individual samples highlighted significant intra-tumoral heterogeneity. These MPs were defined by specific gene modules and enriched pathways (Figs. 2B, C). MP_01 corresponded to a generic mitotic program (G2/M), marked by MKI67 and TOP2A. MP_02, associated with the G1/S transition, featured active DNA replication (CCNE1+PCNA+). MP_03 was linked to stress response and cellular senescence signatures, including DNA-damage-related genes (DDIT3, FOS) and cell cycle arrest genes (CDKN1A, CDKN2A), and was enriched with chemokine genes (CXCL2/3/8), indicative of the senescence-associated secretory (SASP) phenotype [10]. MP_04 exhibited heightened metabolic activities with enzyme-coding genes like ENO1 and LDHA. MP_05 was associated with activated antigen-presentation pathways (HLA-B+HLA-C+). MP_06 was enriched in glandular and mucosal defense genes (e.g., TFF1/2/3, CEACAM5). Lastly, MP_07 presented a distinct interferon (IFN) response program, featuring IFN-stimulated genes (ISGs) such as IFIT1/3, IFITM1/2, and ISG20.
Fig. 2
Decoding shared and distinct malignant meta-programs between CCA subtypes. (A) NMF analysis of mEPCs. Heatmap is colored by Jaccard similarity. Numbers on the top represent seven meta-programs. (B) Heatmap showing gene signatures in seven meta-programs normalized by z-score. Meta-programs are labeled the same as (A). (C) Bubble plot showing enrichment of GO biological processes across seven meta-programs. (D) Violin plots comparing meta-program enrichment scores between different CCA subtypes. (E) Heatmap showing four subtypes of in-house CCA scRNA-seq cohort based on meta-program scores. LN, lymph node; MVI, microvascular invasion. (F-I) Representative IHC staining images of markers for four malignant subtypes of our CCA tissue microarray. (J-M) Kaplan-Meier curves comparing overall survival of four subtypes markers in our CCA tissue microarray cohort. The p values are from Wilcoxon test. *, p < 0.05; **, p < 0.01; ***, p < 0.001
We further explored the distribution of these cell states across CCA subtypes by quantitively assigning MP scores to individual cells. The senescence state (MP_03) was predominant in iCCAs, while the gland (MP_06) and IFN response (MP_07) states were more prevalent in eCCAs (Figs. 2D and S4B-C). We classified each sample into MP-defined subtypes by averaging the expression of corresponding MP signatures. Hierarchical clustering identified four subtypes: proliferative (G1/S and G2/M signatures), glandular (mucosal defense and metabolism signatures), IFN-responsive (glandular and IFN-related signatures), and senescent subtype (Fig. 2E). The IFN-responsive subtype, primarily comprising eCCAs, was associated with advanced stages, lymph node metastasis, and microvascular invasion (Figs. 2E and S4D). We further explored the prognostic significance of molecular subtypes in an independent CCA cohort based on tissue microarray (TMA) data (n = 133; Fig. 2F-M), by using subtype-specific markers (proliferative: MKI67; senescent: ATF3; IFN-responsive: IFITM1; glandular: TFF1). High expression of MKI67 and IFITM1 were both associated with worse prognosis, whereas increased ATF3 expression correlated with better survival (Fig. 2J, L). Validation using the International Cancer Genome Consortium (ICGC) microarray dataset revealed similar patterns of subtype distribution (Fig. S4E-G), with the IFN-responsive subtype enriched in eCCAs and associated with advanced stages and poor prognosis (Figs. S4H, I). Collectively, these findings systematically characterize the transcriptomic heterogeneity between iCCAs and eCCAs, providing insights into CCA molecular subtypes and their clinical relevance.
Expanding the application of molecular subtyping in functional precision oncology with PDOsWe then sought to determine whether molecular subtyping could offer therapeutic insights for precision medicine in CCAs. Given the established value of PDO models in functional precision oncology and their efficiency in large-scale drug screening [11], we leveraged our pre-established CCA PDO library to validate the molecular subtypes. We successfully established sixteen stable, treatment-naïve CCA PDOs (ten iCCAs, five eCCA-Ps, and one eCCA-D). Morphologically, these PDOs displayed thickened walls invaginating into the biliary lumens with tumor-like pleomorphic architectures. Hematoxylin-Eosin (H&E) staining validated that PDOs displayed nuclear atypia, abundant mitotic figures, and pleomorphic nuclei, faithfully preserving the malignant features of corresponding tissues (Fig. 3A). Deconvolution of the PDO bulk RNA-seq data with MP signatures revealed the predominant molecular subtype for each PDO line. Notably, the IFN-response program was enriched in eCCA PDOs, in line with our NMF-based findings (Fig. 3B).
Fig. 3
CCA subtypes recapitulated in PDO models linking to drug response. (A) Representative bright field and H&E staining images of 16 CCA PDOs. (B) Heatmap showing subtyping of 16 PDOs based on NMF meta-program scores. (C) Heatmaps displaying high-throughput screening of 175 drugs across four subtypes of PDOs, with the right and left side heatmap showing cell viability and normalized viability, respectively. Barplots on the left side of heatmaps showing scaled sums of z-scores representing the overall drug response. Drugs are colored by related pathways and are clustered by rows. (D-G) Barplot showing top 30 effective drugs in proliferative-subtype, senescent-subtype, IFN responsive-subtype and glandular-subtype PDOs
We subsequently explored whether different subtypes could predict responses to anti-cancer therapeutics by using a library of 175 compounds, which were systematically grouped at pathway levels (Fig. S5). A proof-of-principle single concentration drug-discovery screening approach (10 µmol/L) was utilized for each compound. Compounds resulting in less than 50% cell viability in PDOs after treatment were considered effective [12]. High-throughput PDO drug screening revealed substantial heterogeneity of drug vulnerabilities in a personalized manner (Fig. S5). We then integrated drug screening profiles of PDOs at the subtype level (Fig. 3C), and selected the top 30 agents with the highest inhibition rate for each PDO subtype (Figs. 3D-G). These agents predominantly targeted pathways related to DNA damage response, protein tyrosine kinases, epigenetic reprogramming, and cytoskeletal signaling, which are among the most extensively studied in BTC treatment [8]. Narrowing our focus on these pathways (Fig. 3C), both in the context of the full drug panel and when analyzing the top 30 compounds, the IFN-responsive subtype consistently displayed significantly greater drug resistance. In contrast, the senescent and glandular subtypes generally exhibited increased drug vulnerability (Figs. 3D-G).
Collectively, PDO models recapitulated and validated the core biological traits and molecular subtyping of CCAs. Pharmacotyping of PDOs further advanced our understanding of phenotypic drug discovery in CCAs [13], and highlighted the potential of molecular subtyping to inform precision therapy.
Identifying eCCA-specific basal-like progenitor subpopulationsBeyond the functional partitioned NMF-based programs, we next asked whether distinct cancer cell subpopulations were unique for CCA subtypes. We identified 17 clusters of malignant EPCs (mEPCs), totaling 12,858 cells (cluster 0–16) (Fig. 4A; Table S5). iCCAs displayed greater inter-tumoral heterogeneity than eCCAs, but no significant difference was revealed between eCCA-Ps and eCCA-Ds (Figs. S6A-B). Several clusters were occupied by individual samples; however, a small cluster (c7) was predominantly composed by eCCAs (Figs. 4B and S6C-D). Differential gene expression (DGE) analysis revealed that c7 highly expressed basal-squamous cell markers (e.g., KRT5, KRT6A, LY6D, KLK7, CDH3; annotated using the Human Protein Atlas), which were also linked to development and stemness [14,15,16] (Figs. 4C-F). CytoTRACE analysis showed the stem-cell-like nature of this cluster (Fig. 4G). Monocle analysis demonstrated that c7 cluster initiated the trajectory and subsequently bifurcated into two branches encompassing other cancer cell lineages presumably accounting for different eCCA subtypes (Fig. 4H-I). Consistently, the expression of LY6D, KLK7, CDH3, CD44, SOX9, and EPCAM decreased along the pseudotime trajectory (Fig. 4J). Pathway analyses indicated enrichment of epidermis development/differentiation and IFN response-associated pathways in the c7 subcluster (Fig. 4K). In agreement, we observed a notably elevated score for the NMF-based IFN-response MP (Fig. S6E), further underscoring the critical role of IFN signaling in this subpopulation. To validate these findings, We leveraged three independent single-cell cohorts comprising 13 eCCAs and 5 adjacent normal tissues (ANTs) (Fig. S7A), including two scRNA-seq datasets [17, 18], and one snRNA-seq dataset [19]. Following data integration and cell annotations (Figs. S7B-E), we identified a basal-like epithelial cluster (Epi_05) characterized by the expression of LY6D and KLK7, which was preferentially enriched in tumor samples (Figs. S7F-I). Meanwhile, Epi_05 highly expressed multiple ISGs particularly ISG15 (Fig. S7G). Using gene signatures based on our IFN-response MP, Epi_05 exhibited the highest IFN-response score compared to other clusters (Fig. S7J).
Fig. 4
Identification of eCCA-specific basal-like progenitor populations. (A) UMAP plot of mEPCs colored by clusters. (B) Pie chart showing the patient composition of cluster 7. (C) Scatter plot displaying upregulated marker genes of cluster 7. (D-F) UMAP density plot showing LY6D, KLK7 and CDH3 expression across mEPCs. (G) UMAP plot showing differential transcriptional entropy of mEPCs calculated by CytoTRACE. (H-I) Pseudotime analysis of mEPCs from eCCA-P and eCCA-D patients. Cells are colored by Seurat clusters and sample types. (J) The dynamic expression of marker genes of cluster 7 along the pseudotime. (K) Bubble plot showing enrichment of GO biological processes using cluster 7 DEGs
We confirmed LY6D+panCK+ cancer cells in eCCA samples via multicolor immunohistochemical (mIHC) staining (Fig. 5A). Employing canonical markers in scRNA-seq dataset to define major cell types (e.g., EPCAM and KRT19 for EPCs), we used RNA in situ sequencing (ISS) to validate the existence of LY6D+ cancer cells (Figs. 5B and S8A-C; Table S6), revealing higher levels of basal-like markers (KLK7, KRT5, KRT10), stem cell markers (CD44, SOX9, EPCAM), and proliferative markers (MKI67, PCNA) in LY6D+ versus LY6D− cancer cells (Fig. 5C). Leveraging in vitro models, we confirmed LY6D expression in the eCCA-P cell line QBC-939. FACS sorting identified LY6D+ cells at a proportion of 6.68% (Fig. 5D). LY6D+ cells exhibited greater spheroid formation capacity, with upregulated stem cell genes (CD44, CD133, EPCAM) and basal cell genes (KLK7, CDH3) (Figs. 5E, F). LY6D+ cells also displayed enhanced resistance to gemcitabine, cisplatin and 5-fluorouracil (5-FU) (Figs. 5G), in line with the greater drug resistance of the IFN-responsive PDO subtype. Subcutaneous engraftment mice models further confirmed that LY6D+ cells significantly generated larger outgrowth of tumors than LY6D− cells (Figs. 5H-I), with upregulated expression of CD44, CD133 and EpCAM in LY6D+ cells-generated tumors (Figs. 5J). Collectively, these results demonstrated that LY6D+ cells behaved as eCCA-specific basal-like progenitors associated with cancer stemness and chemotherapy resistance.
Fig. 5
eCCA-specific basal-like progenitors showing basal cell and stemness features, with increased drug resistance. (A) Multiplex IHC of LY6D+ EPCs in three eCCA patients, indicted by white arrows. DAPI: deep blue. Pan-CK: light yellow. LY6D: pink. Scale bar: 10 μm. (B) RNA in situ sequencing of three eCCA patients. LY6D+ and LY6D− EPCs are labeled by red and blue, respectively. Cell nuclei are labeled by white. (C) RNA in situ sequencing revealing differential expression of basal, stemness and proliferative marker genes between LY6D+ and LY6D− EPCs. (D) Sorting of LY6D+ and LY6D− subpopulations in QBC-939 eCCA cell line by flow cytometry. (E) Bar plot comparing RT-qPCR results of marker genes between LY6D+ and LY6D− subpopulations of the QBC-939 eCCA cell line. (F) Barplot comparing spheroid formation capacity between LY6D+ and LY6D− subpopulations of the QBC-939 eCCA cell line. (G) Dose-response curves showing differential responses to gemcitabine, cisplatin, and 5-fluorouracil between LY6D+ and LY6D− subpopulations of QBC-939 eCCA cell line. (H) Photos comparing subcutaneous tumor growth after injection of LY6D+ and LY6D− populations from the QBC-939 eCCA cell line in nude mice. (I) Bar plot comparing average tumor volumes after injecting LY6D+ or LY6D− cells. (J) Western blot analyses comparing protein expression of LY6D and stemness markers between LY6D+ and LY6D− tumors. Statistical analyses are from t-test. *, p < 0.05; **, p < 0.01; ***, p < 0.001
Unique mesenchymal microenvironment between different CCA subtypesAnother critical question in CCAs, archetypal desmoplastic cancers, is how cancer-associated fibroblasts (CAFs) act as key orchestrater of the TME and contribute to the stromal heterogeneity across CCA subtypes [20]. We identified 18,555 mesenchymal cells, clustering into five fibroblast subsets (c1-c5; LUM+COL1A1+) and three pericyte subsets (c6-c8; RGS5high) (Figs. 6A and S9A-B; Table S7) [21]. Two fibroblast clusters, benign fibroblasts (benign Fib, c1) and inflammatory fibroblasts (iFib, c2), were preferentially enriched in iBTs. The benign_Fib highly expressed genes involved in extracellular matrix (ECM) development and remodeling, including MFAP5 and IGFBP6 [22, 23]. The iFib cluster exhibited elevated expression of inflammatory markers such as C3, C7, and CCL2, alongside activation of pathways associated with leukocyte chemotaxis and immune responses (Figs. 6B-E). The remaining three fibroblast subsets, enriched in CCAs, were classified as CAFs (Figs. 6A-C). Matrix CAFs (c3) expressed MMP11 and CST1 which facilitated tumor invasiveness and metastasis [24]. Stem-like CAFs (c4) were marked by stem cell markers such as GPC3 and SOX9. Senescent CAFs (senCAFs, c5), chiefly residing in iCCA specimens, expressed senescence markers (CDKN2A, SERPINE1) (Figs. 6A-D) [25]. SenCAFs uniquely expressed MCT4 and PKM2, both participating in glycolytic reprogramming during hepatic stellate cells (HSCs) activation and transdifferentiation towards a myofibroblast phenotype [26]. Pathway analyses validated the functional specialization of these clusters (Figs. 6E and S9C). Evidenced by the CytoTRACE transcriptional entropy, stem-like CAFs were at a less differentiated state, whereas senCAFs were oppositely at a more differentiated state (Figs. S9D, E).
Fig. 6
scRNA-seq analysis of CCA mesenchymal microenvironment. (A) UMAP showing the mesenchymal cells colored by cell subtypes. iFib, inflammatory fibroblast; senCAF, senescent CAF; iPer, inflammatory pericyte; myoPer, myofibroblast pericyte. (B) UMAP showing mesenchymal cells colored by sample types. (C) Sample preference of the mesenchymal subtypes analyzed by Ro/e. (D) Heatmap showing average expression of marker genes across eight mesenchymal subtypes. (E) Barplots showing enrichment of GO biological processes across eight mesenchymal subtypes. (F) Pie chart displaying the percentage contribution of sample sources in ISG15 Per. (G) Bubble plot showing cell-cell interaction of EDN signaling pathways between eCCA mEPC subclusters and Per_ISG15
Uniquely expressing pro-inflammatory markers (CCL19, CCL21, IL6), c6 was defined as inflammatory pericytes (iPer). Myopericytes (myoPer, c7) highly expressed MYH11 and ACTA2, associated with smooth muscle contraction. Both iPer and myoPer clusters were enriched in eCCA-Ps. We also identified a subset of pericytes (c8; Per_ ISG15 highly expressing interferon-stimulated genes (ISGs) such as ISG15, IFI27, and IFITM1/3, predominantly enriched in eCCA-Ps and eCCA-Ds (Figs. 6B-F). ISGs like ISG15 operate both intracellularly and extracellularly, acting as central players in IFN-associated pathways and vividly modulating the host damage and repair response [27]. Cell-cell communication analyses revealed robust interaction between Per_ ISG15 and c7 mEPC cluster, primarily mediated by EDN1 and EDNRA/B ligand-receptor interaction (Fig. 6G).
Immune landscape of CCAs revealing eCCA-specific ISG15 high subsetsImmune cell profiles (e.g., macrophages, T cells) surrounding bile ducts likely establish site-specific immune compartmentalization that dictate localized immune surveillance, yet remain largely uncharted [28]. We firstly identified two monocyte clusters (S100A8+S100A9+), seven macrophage clusters (CD68+CD163+), and four dendritic cell clusters (FCER1A+HLA-DQA1+) (Figs. 7A, B and S10A; Table S8) with differential distributions and functional phenotypes (Figs. 7C-E). Akin to our findings in gallbladder cancer [29], we observed an enrichment of myeloid-derived suppressor cell (MDSC)-like monocytes in iBTs. M2-like macrophages and tumor-associated macrophages (TAMs) were more prevalent in CCAs. The Macro_ISG15 cluster highly expressed ISGs (e.g., ISG15, IFI27) and was preferentially enriched in eCCA-Ds (Figs. 7C and F). Deconvolution of the ICGC microarray dataset using Macro_ISG15 signatures showed that higher expression correlated with shortened survival in eCCAs but not in iCCAs (Figs. 7G and S10C). Cell-cell communication analysis revealed active interaction between the c7 mEPC cluster and Macro_ISG15 via the GAS6-AXL signaling pathway (Fig. S10B).
Fig. 7
scRNA-seq revealing IFN-signaling activated immune subpopulations. (A) UMAP of myeloid cells colored by subtypes. Mono, monocytes; Macro, macrophages; DC, dendritic cells. (B) Bubble plots showing the expression of marker genes across myeloid cell subtypes. (C) Sample preference of myeloid cells subtypes analyzed by Ro/e. (D) Heatmap showing functional enrichment scores across monocyte and macrophage subtypes. TAM, tumor-associated macrophage. (E) Heatmap showing functional enrichment scores across different sample types. (F) Pie chart displaying the percentage contribution of sample sources in Macro_ISG15. (G) Kaplan-Meier curves of overall survival in ICGC-eCCA cohort patients grouped by Macro_ISG15 signature score. (H) UMAP visualization of lymphocytes colored by subtypes. (I) Bubble plot showing the expression of marker genes of lymphocyte subtypes. (J) Sample preference of lymphocyte subtypes analyzed by Ro/e. (K, L) Heatmap showing functional enrichment scores of CD4+ T cell and CD8+ T cell subtypes. (M) Pie chart displaying the percentage contribution of sample sources in CD8T_ISG15. (N) Kaplan-Meier curves of overall survival in ICGC-eCCA cohort patients grouped by CD8T_ISG15 signature score
We further characterized three CD4+ T cell clusters, five CD8+ T cell clusters, two NKT clusters, and one NK cluster (Figs. 7H and S10D). We annotated the CD4T_IL7R cluster as cytotoxic effector CD4+ T cells, the CD4T_CXCL13 cluster as exhausted T cells, and the CD4T_FOXP3 cluster as regulatory T cells (Fig. 7K). Similarly, we characterized naïve T cells (CD8T_IL7R), cytotoxic T cells (CD8T_GZMK), exhausted T cells (CD8T_LAG3), and tissue-resident memory T cells (CD8T_ZNF683) (Fig. 7L). Notably, we identified a subcluster of CD8+ cells (CD8T_ISG15) highly expressing ISG15, IFIT3 and IFI44L (Fig. 7I), enriched in eCCAs (Figs. 7J and M), and showed enrichment of IFN response pathways (Fig. 7L). Deconvolution of the ICGC dataset revealed that elevated expression of CD8T_ISG15 signatures portended worse prognosis for eCCAs but not for iCCAs (Figs. 7N and S10E).
Validation of specific mesenchymal and immune niches for basal-like progenitorsThe data presented above revealed a convergence of specific epithelial, mesenchymal, and immune cell subpopulations (Clusters of mEPC_c7, Per_ISG15, Macro_ISG15, and CD8T_ISG15) surrounding eCCAs (Fig. 8A), which prompted a deeper exploration of their interconnected associations. All of these clusters consistently exhibited elevated expression of multiple ISGs, particularly ISG15 (Fig. 8B), in line with the notion that ISG15 is one of the most rapidly and strongly induced ISGs [27]. Analysis of the public ICGC dataset confirmed higher expression of ISG15 in eCCAs compared to iCCAs (Fig. 8C). In eCCA patients, ISG15 expression correlated strongly with LY6D+ mEPC signatures (e.g., LY6D, KLK7, KRT5, CDH3) and was associated with worse prognosis (Figs. 8D-E). By examining the aforementioned external single-cell datasets of eCCAs, we validated the presence of ISG15high cell clusters across pericytes, myeloid cells, and T/NK cells (Figs. S11A-I). Leveraging annotated markers of major cell types from scRNA-seq, we identified spatial co-location patterns between LY6D+ EPCs and ISG15high pericytes, ISG15high CD8+ T cells, and ISG15high macrophages in RNA ISS experiments (Figs. 8F and S12A). Among the three ISG15high TME subsets, Per_ISG15 cells were found to reside in significantly closer proximity to LY6D⁺ EPCs, suggesting a preferential spatial relationship (Fig. S12B).
Fig. 8
Specific mesenchymal and immune niches sustaining basal-like progenitors. (A) Circular plot displaying the distribution of sample sources for Macro_ISG15, CD8T_ISG15, Per_ISG15 and mEPC_c7: patient sources (left) and sample type origins (right). (B) Heatmap showing average expression of ISG15 and representative upstream genes across mesenchymal and immune cell subtypes. (C) Boxplot comparing ISG15 expression levels between iCCAs and eCCAs in the ICGC cohort. (D) Correlation heatmap analyzing expression of ISG15 and representative DEGs of c7 mEPC clusters in ICGC eCCA patients. (E) Kaplan-Meier curves of overall survival in eCCA patients stratified by ISG15 expression in the ICGC-eCCA dataset. (F) Spatial RNA ISS visualization of LY6D+ EPCs and ISG15-expressing pericytes, macrophages and T cells. LY6D+ EPCs: red. Pericytes: light brown, Macrophages: light blue. T cells: grey. ISG15: green cross. (G, H) Spheroid formation capacity analysis between vehicle- and ISG15 (100 ng/mL)-treated QBC-939 eCCA cell line. (I) Bar plot comparing RT-qPCR results of marker genes between vehicle- and ISG15-treated QBC-939 eCCA cell line. (J) WB analysis comparing proteins levels of LY6D, KLK7 and stemness markers between vehicle- and ISG15 (100 ng/mL)-treated QBC-939 eCCA cell line. Statistical analyses are obtained from t-test. *, p < 0.05; **, p < 0.01; ***, p < 0.001
Given the progenitor-like property of LY6D+KLK7+ mEPCs, we hypothesized that ISG15 might play a role in sustaining this feature. Using the tumor spheroid assay with the eCCA-P cell line QBC-939, we confirmed that ISG15 stimulation significantly increased the spheroid formation capacity of cancer cells (Figs. 8G, H). Quantitative real-time PCR and western blotting (WB) validated the upregulation of basal cell markers (LY6D, KLK7) and stem cell markers (CD44, CD133, EPCAM) (Figs. 8I, J). To further elucidate the role of ISG15 in functional precision oncology, we introduced ISG15 into eCCA PDO and cell line models, and evaluated the vulnerability of cisplatin, gemcitabine, and 5-FU. ISG15 markedly enhanced resistance of cisplatin and gemcitabine in the PDO line, and to cisplatin and 5-FU in the cell line (Fig. S13A). Moreover, using an IFN-responsive PDO line, we proved that the addition of ISG15 could substantially increase resistance to most tested compounds, including those targeting DNA damage, epigenetic reprogramming, or cytoskeletal signaling (Fig. S13B), reminiscent of the drug-resistant features of IFN-responsive PDOs (Fig. 3C).
Collectively, we identified a subpopulation of LY6D+ basal-like progenitors uniquely localized within eCCAs, likely sustained by ISG15high mesenchymal and immune niches. ISG15 is crucial for maintaining cancer cell stemness and augmenting resistance to various anti-cancer agents.
Comments (0)