
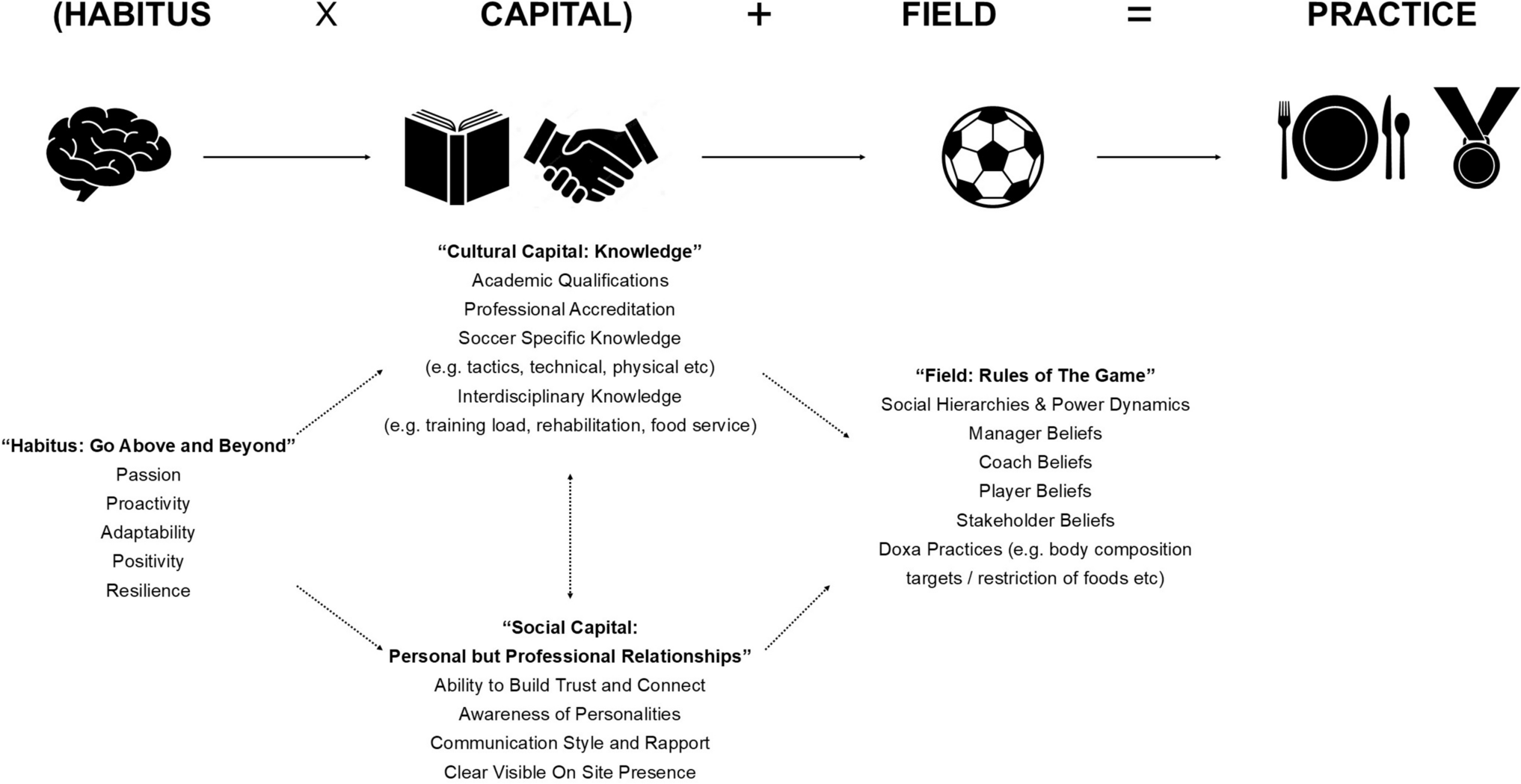
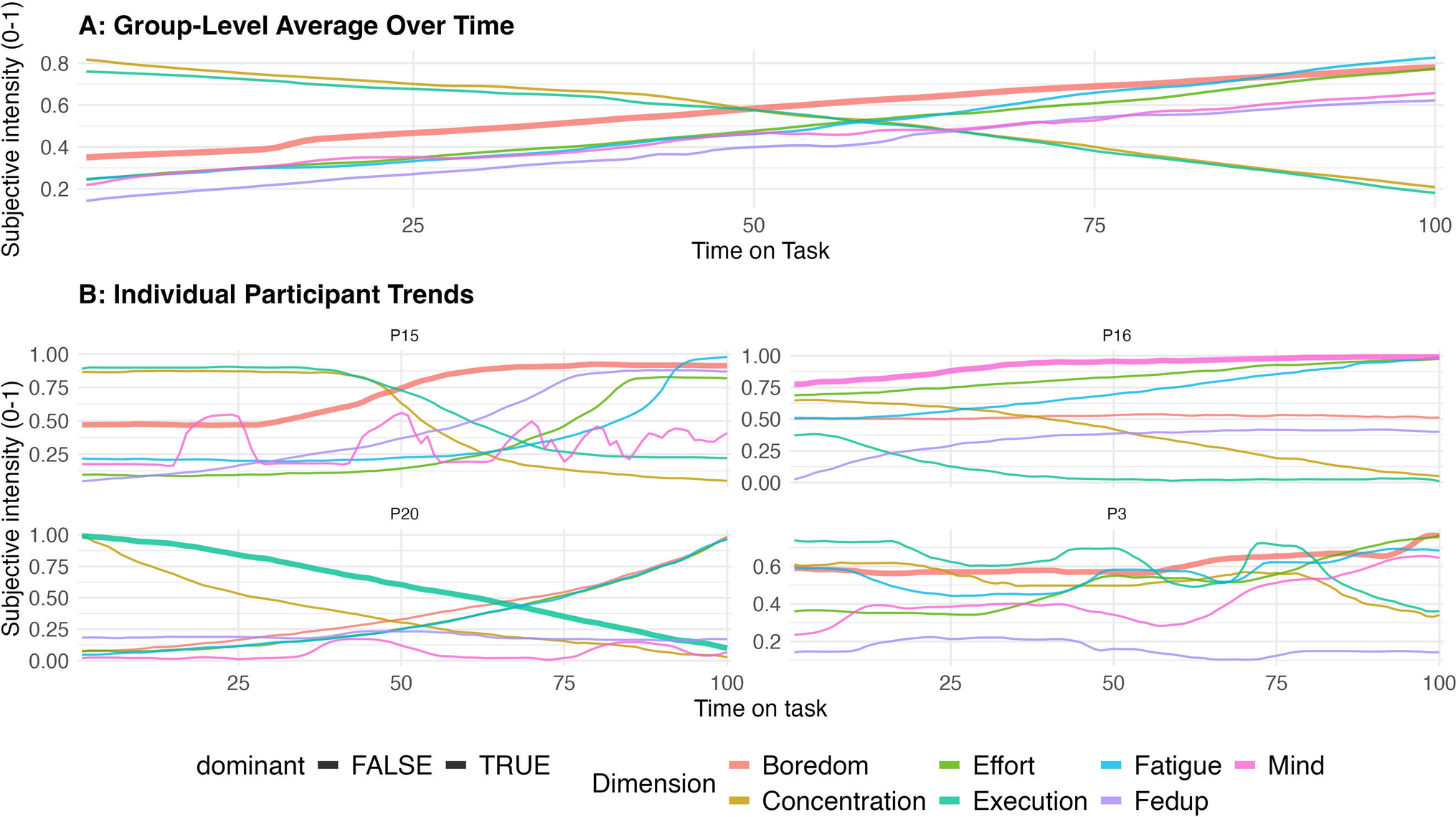
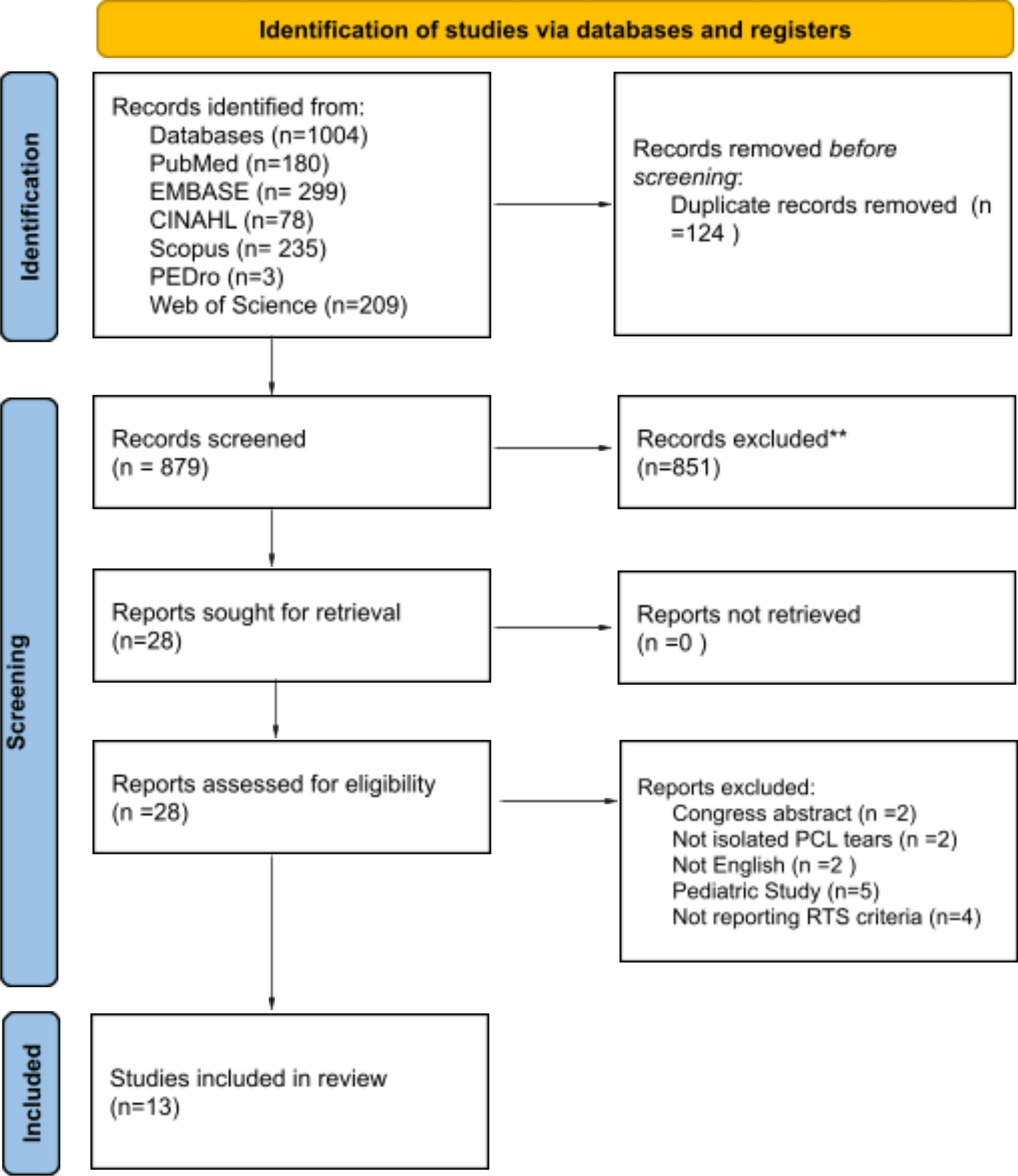
Remember me
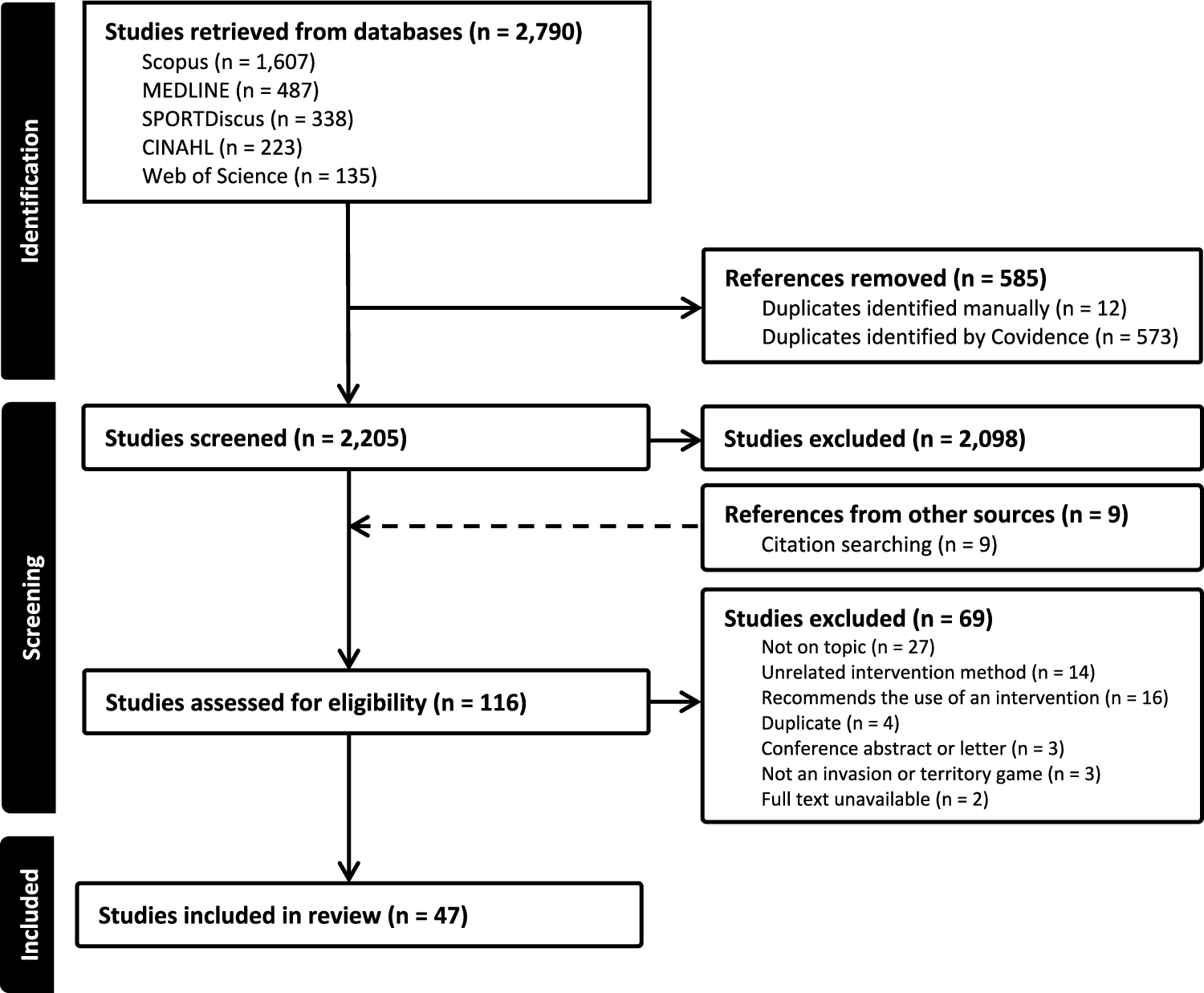
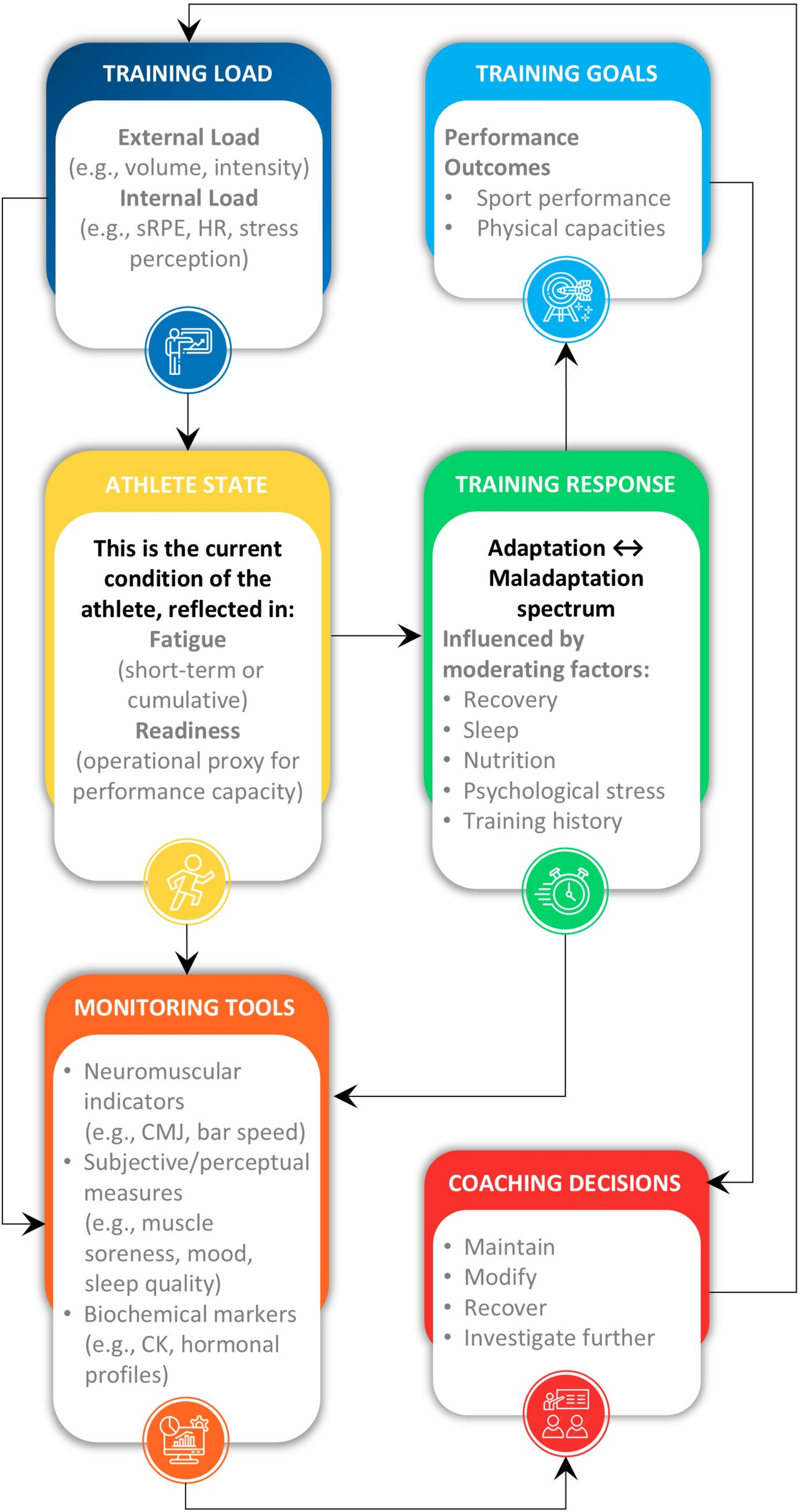
This double-blind, placebo-controlled parallel-group study was conducted within a residential training camp environment where athletes were supervised for all meals and training sessions. The study consisted of a 5-week camp including a 3-week structured altitude training block (Fig. 1). During week 1, athletes were housed at the Australian Institute of Sport (Canberra, ACT, Australia), where they undertook a battery of tests to identify baseline characteristics including submaximal running economy and \(\dot}_}}}\), body composition (assessed via dual-energy X-ray absorptiometry, DXA), and hemoglobin mass (Hbmass) and had a resting venous blood sample taken. Participants in Cohort A completed a 3000 m time trial (TT) on a synthetic rubberized track (n = 22 total; n = 11 UA, n = 11 placebo; PL), while Cohort B (n = 20; n = 11 UA, n = 9 PL) had skeletal muscle biopsies taken to determine the impact of UA supplementation on global protein expression and in situ mitochondrial respiration in permeabilized muscle fiber bundles (PmFB) using high-resolution respirometry. All testing during this period was performed while following a standardized diet of high energy and carbohydrate (CHO) availability (220 kJ·day−1, 8.5 g·kg−1 body mass·day−1 CHO). Upon completion of baseline testing, athletes within each cohort (i.e., Cohort A and Cohort B) were pair-matched on the basis of \(\dot}_}}}\), lifetime running personal bests, and training volume and randomized using a random number generator in a 1:1 ratio to receive either 1000 mg·day−1 UA or placebo (PL; composed of the same inert excipients as the active treatment) for 4 weeks. Pair-matching was done to limit differences between groups (UA versus PL) at baseline. Randomization was performed by a clinical trial manager external to the research team. Both the UA and PL supplements were provided in identical packaging containing visually identical soft gel capsules. Participants consumed 4 × 250 mg capsules (1000 mg total) each morning on an empty stomach with a glass of water. During the 3-week structured training camp, participants were housed at 1800 m elevation (Perisher Valley, NSW, Australia) with training sessions performed at low-to-moderate altitudes (1700–2200 m, Snowy Mountains, Australia).
Fig. 1
Study schematic. Forty-two highly trained male distance runners participated in this 5-week double-blind placebo-controlled clinical trial. Athletes were randomized to receive either 1000 mg·day−1 urolithin A (UA, n = 22) or placebo (PL, n = 20) for 4 weeks while completing an intensified 3-week training camp performed at low altitude (~ 1700–2200 m)
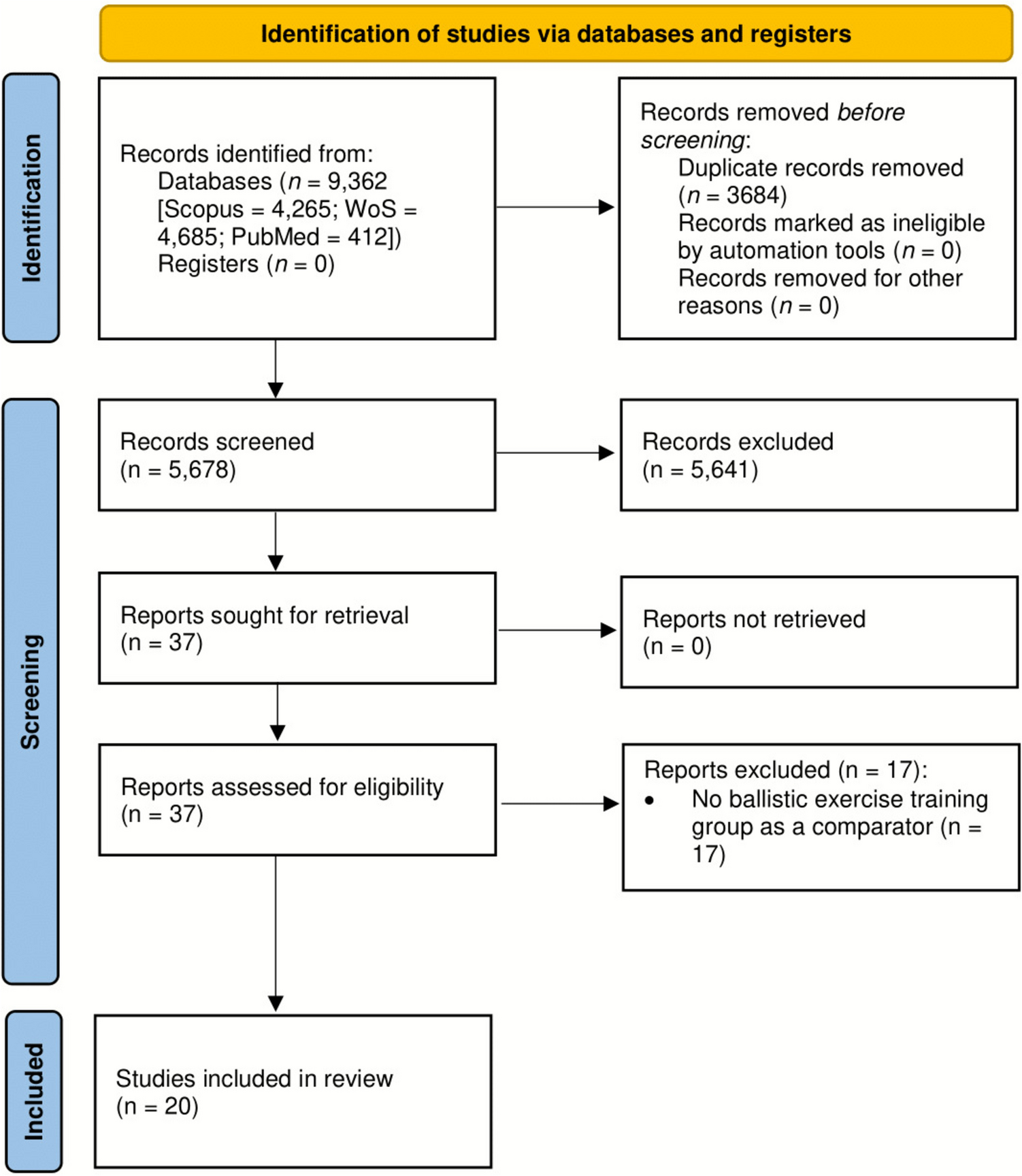
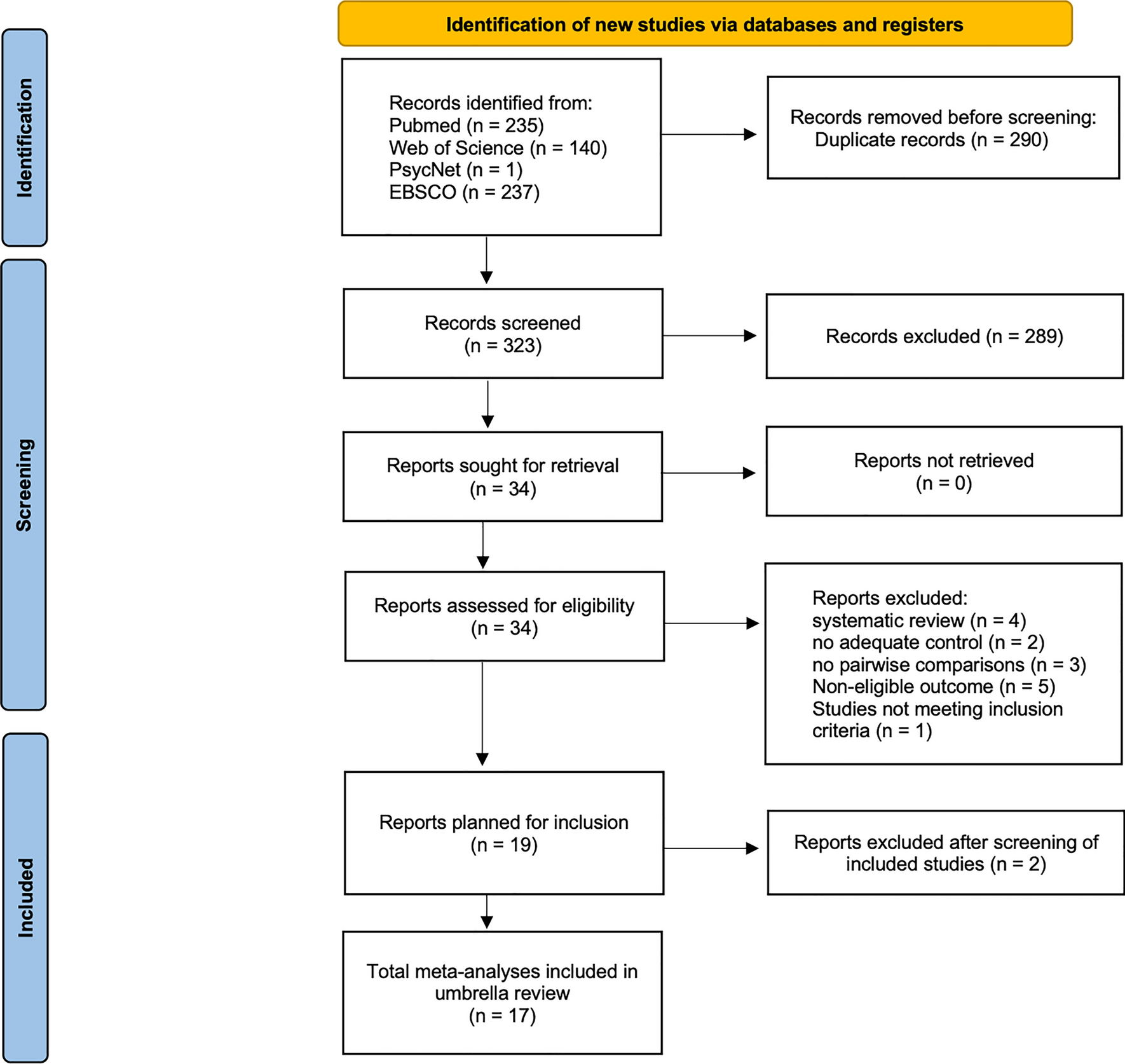
2.2 ParticipantsA total of 44 highly trained (tier 3–4 [21]) male middle- and long-distance runners were recruited between March 2021 and October 2022 to participate in this study. To be eligible, participants were required to be currently running > 70 km·week−1, have a \(\dot}_}}}\) > 60 mL·kg−1·min−1 and have an official 3000 m personal best time of less than 9:00 (Cohort A; performance arm) or 10:00 min (Cohort B; muscle biopsy arm), and agree to participate in the training camp. One participant was lost to follow-up (did not report for baseline testing), and one participant withdrew with severe illness partway through the investigation, unrelated to the treatment. Their data were not included in final analysis, leaving a final sample size of 42 (CONSORT diagram, Supplementary Fig. 1). Subject characteristics are presented in Table 1. Ethics approval was obtained from the Australian Catholic University’s Human Research Ethics Committee (2021-36HC), and the study was prospectively registered as a clinical trial (NCT04783207). Comprehensive details of the study protocol were explained orally and provided in writing prior to athletes providing their written informed consent. All procedures conformed to the standards set by the Declaration of Helsinki.
Table 1 Subject characteristics2.3 Assessments2.3.1 Body CompositionAt baseline and following the 3-week altitude camp, all athletes undertook a DXA assessment for an estimation of body composition (iDXA, GE Healthcare, Milwaukee, WI). These measurements were undertaken according to the Best Practice Protocols of the Australian Institute of Sport; participants reported in the early morning in an overnight fasted and rested state, while the same DXA technician positioned participants and analyzed all images (enCore v18, GE Healthcare). The test–retest technical error of measurement for the iDXA at our center is 0.65% for total mass, 0.95% for lean mass, 1.82% for fat mass, and 0.47% for bone mass.
2.3.2 Hemoglobin MassThe erythropoietic response to altitude exposure and UA supplementation was assessed via measurement of total hemoglobin mass (Hbmass) using the optimized 2-min carbon monoxide (CO) rebreathing method. Briefly, a CO bolus (1.2 mL·kg−1 body mass) was rebreathed with 4 L 100% oxygen through a glass spirometer for 2 min. Carboxyhemoglobin (HbCO) concentration of capillary blood was measured in quintuplet before and 7 min post CO ingestion using an OSM 3 hemoximeter (Radiometer, Copenhagen, Denmark). Hbmass was calculated from the mean change in HbCO as described previously [22]. Hbmass was assessed at baseline and following the completion of the 3-week altitude training camp.
2.3.3 Incremental Exercise TestingPrior to and following the altitude camp, all participants completed an incremental exercise test to exhaustion on a custom-built motorized treadmill (Australian Institute of Sport, Bruce, Australia) to determine submaximal running economy and \(\dot}_}}}\). This test commenced 2 h after the intake of a standardized meal providing 2 g·kg−1 body mass CHO. A self-selected warm-up of 10 min duration preceded the test, which was maintained across trials. Running economy was assessed during four submaximal stages, each lasting 4 min and increasing in speed by 1 km·h−1 followed by 1 min standing rest. Starting speeds were selected at either 14 (Cohort B) or 16 km·h−1 (Cohort A) on the basis of each individual’s capacity, increasing to 17 or 19 km·h−1 at the final stage. Heart rate (HR) was measured continuously throughout the test (Polar Heart Rate Monitor, Polar Electro, Kempele, Finland). Expired gas was collected and analyzed using a custom-built indirect calorimetry system [14], with the final 60 s of gas collected accepted as steady state and used to calculate the respiratory exchange ratio (RER) and O2 uptake. Upon completion of the final submaximal stage, participants rested for 5 min before completing a ramp (speed and then gradient) test to volitional fatigue. The incremental test commenced at either 13 (Cohort B) or 15 km·h−1 (Cohort A) and was increased by 0.5 km·h−1 every 30 s until the speed corresponding to the individual’s final submaximal stage was reached (17 or 19 km·h−1), with treadmill gradient increased by 0.5% every 30 s thereafter until exhaustion. Expired gas was collected and analyzed throughout, and maximal HR recorded upon completion of the test. RER was calculated from steady-state expired gases collected over 1-min periods during the submaximal economy and \(\dot}_}}}\) protocol. Rates of CHO and fat oxidation (g·min−1) were calculated as described previously [23]. Briefly, \(\dot}_}\) and \(\dot}_}\) values were used to calculate substrate oxidation rates using nonprotein RER values [24]. These equations are based on the premise that \(\dot}_}\) and \(\dot}_}\) accurately reflect tissue O2 consumption and CO2 production, and that indirect calorimetry is a valid method for quantifying rates of substrate oxidation in well-trained athletes during strenuous exercise of up to 85% of \(\dot}_}}}\) [25].
2.3.4 3000 m Race PerformancePrior to (Race 1) and following (Race 2) the altitude camp, participants in Cohort A completed a 3000 m time trial (TT) on a synthetic 400-m outdoor athletics track (Canberra, ACT, Australia). Race performance was individually hand-timed for each participant by a member of the research team, with elapsed time for each lap being announced during the race. Each race was preceded by a self-selected warm-up of ~15 min duration, which was maintained across trials for each participant. Capillary blood samples were collected immediately prior to and following the completion of the race for blood lactate analysis, with RPE (Borg Scale, 6–20) assessed at the same time points. Additional samples were collected for analysis of circulating markers of inflammation and muscle damage as outlined below.
2.3.5 Downhill Running BoutTo induce muscle damage, participants completed a bout of downhill running on Monday morning of each week at the altitude camp. The run consisted of a self-selected warm-up that was replicated across trials, followed by a ~4.5-km run on an asphalt surface with an average decline of 4%. Participants were instructed to run this as hard as possible and upon completion were driven back to the athlete residence for collection of capillary blood samples for assessment of inflammation and muscle damage markers as outlined below. Training in the day prior to and following the completion of the downhill run was replicated each week.
2.3.6 Circulating Markers of Inflammation and Muscle DamageCapillary blood samples were collected from each participant to assess both CK and CRP in response to the 3000 m TT and the downhill runs. For the TT, samples were collected prior to the warm-up (0 h), as well as 1 and 24 h post race. For downhill runs, samples were collected in the morning prior to commencing exercise (0 h), as well as 1, 24, and 36 h post exercise. Blood collection required athletes to place a hand in a bucket of warm water for ~5 min to increase blood circulation, after which the hand was dried, and an incision made into a fingertip using a lancet. A 1 mL capillary blood sample was collected into a serum separator tube (Minicollect, Greiner Vacuette, Austria), which was left to clot for 30 min before being centrifuged at 1500 g for 10 min. Serum was then divided into cryotubes and frozen at −80°C for batch analysis. CK and CRP were analyzed using a COBAS Integra 400 automated biochemistry analyzer (Roche Diagnostics, Rotkreuz, Switzerland).
2.3.7 Skeletal Muscle Biopsy CollectionParticipants in Cohort B arrived at the laboratory following the consumption of a standardized meal having abstained from caffeine, alcohol, and exercise for the preceding 24 h. Local anesthetic (1% lignocaine hydrochloride in saline; McFarlane; Surrey Hills, Victoria Australia; 11037-AS) was administered to the vastus lateralis, after which two percutaneous skeletal muscle biopsies were collected using a Bergstrom needle modified with suction. A portion of the first biopsy was used for the preparation of permeabilized muscle fiber bundles (see below), while the second sample was immediately snap-frozen in liquid nitrogen and stored at −80°C for subsequent analysis.
2.3.8 Preparation of Permeabilized FibersA small portion of each biopsy was placed in ice-cold BIOPS (50 mM MES, 7.23 mM K2EGTA, 2.77 mM CaK2EGTA, 20 mM imidazole, 0.5 mM DTT, 20 mM taurine, 5.77 mM ATP, 15 mM PCr, and 6.56 mM MgCl2·H2O; pH 7.1) and separated under a microscope into bundles using fine-tipped forceps as described previously [26]. Fiber bundles were then treated with 30 μg·mL−1 saponin for 30 min at 4°C, then washed for 15 min in MiR05 respiration buffer (0.5 mM EGTA, 10 mM KH2·PO4, 110 mM sucrose, and 1 mg·mL−1 fatty acid-free bovine serum albumin (BSA); pH 7.1). Measurements of O2 consumption were performed using high-resolution respirometry (Oxygraph-2K, Oroboros Instruments, Innsbruck, Austria) at 37°C in the presence of 25 μM blebbistatin as previously described [27]. Pyruvate supported respiration was initiated with 10 mM pyruvate and 2 mM malate (PM), followed by 5 mM ADP (+ ADP), 10 mM glutamate (+ G), and 10 mM succinate (+ S). Lipid supported respiration was initiated with 0.2 mM octanoyl-carnitine and 0.5 mM malate (+O-Carnitine), followed by 2.5 mM ADP (+ ADP), and 10 mM succinate (+ S). Both protocols were concluded by adding 10 μM cytochrome c to assess mitochondrial membrane integrity. Experiments with a cytochrome c response greater than 10% were omitted from final analysis.
2.3.9 Muscle Proteomics Sample PreparationHuman muscle samples were homogenized and denatured using a urea-based proprietary denaturing buffer (Biognosys’ Denature Buffer). Samples were homogenized in Biognosys’ SDS Lysis Buffer using a Precellys Evolution homogenizer. Lysates were further prepared on a Hamilton Microlab STAR liquid handling system according to Biognosys’ standard operating procedures. Protein concentrations were measured with a BCA assay (Pierce, Thermo Fisher). Per sample, 70 µg of protein were reduced, alkylated, and digested to peptides using trypsin (Promega, 1:50 protease to total protein ratio) at 37°C. Peptides were desalted using an HLB µElution plate (Waters) and dried down. Peptides were resuspended in 1% acetonitrile/0.1% formic acid in water and spiked with Biognosys’ iRT kit calibration peptides. Peptide concentrations were determined with a microBCA assay (Pierce, Thermo Fisher).
For DIA LC–MS/MS measurements, 3.85 µg of peptides per sample were injected on an in-house packed reversed phase column on a ThermoScientific Vanquish Neo UHPLC nano-liquid chromatography system connected to a ThermoScientific Orbitrap Exploris 480 mass spectrometer equipped with a NanosprayFlex ion source and a FAIMS Pro ion mobility device (ThermoScientific). LC solvents were A: water with 0.1% FA; B: 80% acetonitrile, 0.1% FA in water. The nonlinear LC gradient was 1–50% solvent B for 172 min followed by a column washing step at 90% B for 5 min, and a final equilibration step of 1% B for one column volume at 64°C with a flow rate set to ramp from 500 to 250 nL·min−1 (min 0: 500 nL/min, min 172: 250 nL/min, washing at 500 nL/min). The FAIMS-DIA method consisted per applied compensation voltage of one full range MS1 scan and 34 DIA segments as described previously [28]. For whole-proteome analysis, DIA mass spectrometric data were analyzed using the software Spectronaut (version 17.1, Biognosys) with the default settings, including a 1% false discovery rate control at PSM, peptide, and protein level, allowing for two missed cleavages and variable modifications (N term acetylation, methionine and proline oxidation, ammonia loss, deamidation (NQ)). A human UniProt fasta database (Homo sapiens, 2023 01 01) was used, the default settings were used for the library generation. HRM mass spectrometric data were analyzed using Spectronaut software (Biognosys, version 17.1). The false discovery rate on peptide and protein level was set to 1% on experiment level, and data were filtered using row-based extraction. The direct DIA spectral library generated in this project was used for the analysis. The HRM measurements analyzed with Spectronaut were adjusted using global normalization to median intensity of proteins identified in all runs (sparse).
2.3.10 Differential Protein Expression AnalysisFor testing of differential protein abundance comparing baseline to post-treatment for each group, protein intensities for each protein were analyzed using a two-sample Student’s t test (simple model). To assess protein expression changes between baseline and post-UA treatment visits while accounting for placebo effects, a linear mixed model framework was employed using the msqrob2 package [29]. Raw protein intensity values underwent log transformation followed by normalization using the median-center method. The model was formulated as follows: treatment + visit + treatment:visit, where treatment represents either UA or placebo, and visit indicates baseline or post-treatment visit. Visit was treated as an interaction term in the model. To assess the mean log2 expression between post-UA and baseline, corrected for Placebo effects, the contrast “(Intercept) + post + UA:post” was employed, with the respective statistical test being “post + UA:post = 0.” Volcano plots were generated using − log10 of the nominal p-value (y-axis) and log2 fold change (x-axis) from the comparison of post-UA treatment versus baseline controlling for PL. Proteins with an absolute log2 fold change greater than 0.25 and a nominal p value less than 0.05 were considered significantly regulated. The top 10 proteins by [rank | p value | log2 fold change] were then labeled.
2.3.11 Gene Set Enrichment AnalysisFor gene set enrichment analysis (GSEA), the cellular components (CC) collection of the Gene Ontology database (GO CCs) was utilized for both simple and linear mixed models. Enrichment analysis was performed separately for the “simple” and “robust regression” models using the R package ClusterProfiler [30]. Log2 fold change values between visits served as the protein ranking metric. Subsequently, the enrichment of GO CC gene sets among up- or downregulated proteins was statistically tested. The minimum and maximum gene set sizes were set to 10 and 500, respectively. Adjusted p values were determined using previously described methods [31]. Terms with an adjusted p value ≤ 0.05 were considered statistically significant and further characterized based on the respective normalized enrichment score (NES) sign as either activated (NES > 0) or repressed (NES < 0). Visualizations included Venn diagrams to depict common and unique activated/repressed GSEA GO CCs between placebo and UA comparisons, dot plots of protein ratio, and paired boxplots of normalized protein expression values, highlighting top enrichment proteins within specific terms, specifically the “mitochondrial protein-containing complex” GO CC term in the “robust regression” analysis.
2.3.12 ImmunoblottingMuscle tissues were lysed in denaturing buffer as described above with added protease and phosphatase inhibitor cocktails (Thermofisher, Waltham, MA, USA). Samples were sonicated for 20 min using an ultrasonic bath (Branson 1510) and centrifuged at 13000 RPM for 20 min at 4°C. Protein concentrations from collected supernatants were determined via DC protein assay (Bio-Rad Hercules, CA, United States, 500-0112). Lysates were eluted in 5 × Laemmli buffer (Bio-Rad, 1610747), and 10 μg of each sample was separated by SDS-PAGE (Bio-Rad, 4568086) and transferred onto PVDF membranes (Bio-Rad, 1704156). Membranes were washed in Tris-buffered saline containing 0.05% Tween 20 (TBS-T) and blocked for 1 h with 5% bovine serum albumin (Panobiotech, PANP06-1391100). The following primary antibodies were incubated overnight diluted at 4°C diluted in blocking buffer: Phospho-Parkin (Biorbyt, orb312554, 1:1000), total Parkin (Santacruz, sc-32282, 1:1000), OXPHOS Antibody Cocktail (Abcam, ab110413, 1:2000), and total 4-hydroxynonenal (Abcam, ab46545, 1:1000). After washing with TBS-T, membranes were incubated with secondary antibody (goat–anti-mouse, Abcam 10461444 and goat–anti-rabbit, Abcam 1051204-4, 1:10,000) for 1 h at room temperature. Membrane proteins were detected by enhanced chemiluminescence and the Chemidoc MP imaging system (Bio-Rad). The volume density of each target band was quantified using Bio-Rad Image Lab and normalized to total protein in each lane using stain-free imaging technology using Image Lab software (version 6.1, Bio-Rad), as previously described [32].
2.3.13 Citrate Synthase ActivityMuscle samples were lysed in homogenization buffer containing 50 mM Tris–HCl (pH 7.5), 1 mM EDTA, 1 mM EGTA, 10% glycerol, 1% Triton-X, 50 mM sodium fluoride, 5 mM sodium pyrophosphate with cOmplete Protease Inhibitor Cocktail, and PhosSTOP phosphatase inhibitor (Sigma-Aldrich, St. Louis, MO, USA). Samples were centrifuged at 16,000 g for 30 min at 4°C and protein concentration was determined in triplicate via bicinchoninic acid protein assay (Pierce, Rockford, IL, USA), against bovine serum albumin standards (Sigma–Aldrich). Samples were freeze-thawed 3 × in liquid nitrogen to disrupt cellular membranes prior to use in the assay, and then centrifuged at 900 g for 10 min at 4 °C. Citrate synthase (CS) activity was determined in triplicate on a microplate by adding the following: 5 μL of muscle homogenate at a concentration of 2 µg·µL−1, 40 μL of 3 mM acetyl coenzyme A, and 25 μL of 1 mM 5,5′-dithiobis(2-nitrobenzoic acid) to 165 μL of 100 mM Tris buffer (pH 8.3) kept at 30°C. After the addition of 15 μL of 10 mM oxaloacetic acid, the plate was immediately placed in a plate reader (SpectraMax Paradigm, Molecular Devices, Sunnyvale, CA, USA) at 30°C. Absorbance was read at 412 nm and was recorded every 15 s for 3 min after 30 s of linear agitation. CS activity was normalized to protein content determined via BCA assay and is reported as µmol·min−1·g−1 protein.
2.3.14 Evaluation of Urolithin A PharmacokineticsPharmacokinetics of the main urolithin A metabolite UA-glucuronide were analyzed in a subset of participants (n = 6 UA, n = 4 PL) using dried blot spots (DBS) collected on blood collection cards (Whatman Filter Paper 903 spots card). Fingertip capillary blood samples were collected prior to consuming the first dose of either UA or PL (0 h), and then at 1, 2, 6, and 24 h post ingestion. The 24 h timepoint was collected prior to the ingestion of the subsequent dose of either UA or PL. Each sample consisted of three to four blood spots containing ~ 20–40 µL of whole blood that were spotted on DBS cards. The cards were subsequently dried and stored in sealable biohazard foil bags containing desiccant at room temperature until analysis. Analysis and quantification of UA glucuronide was performed using liquid chromatography coupled to mass spectrometry as described previously [33]. The quantification of UA glucuronide was performed by column separation with reversed phase liquid chromatography followed by detection with triple-stage quadrupole MS/MS in the selected reaction monitoring mode. The concentration of UA glucuronide was calculated using the internal standardization method, with a quantification limit of 5.00–5000 ng mL−1. The acquisition and processing of data was performed using LCquan version 2.5.6 and Xcalibur version 2.0.7 (Thermo Fisher Scientific).
2.4 Statistical AnalysisTo account for two primary endpoints (CK for muscle damage and 3000 m TT performance), a hierarchical order for testing null hypotheses was selected a priori in the study protocol, including a clear specification of the set of hypotheses that need to be significant [34]. For this study, a change in CK was tested first and then race performance was subsequently tested as a co-primary endpoint. The 95% CIs for treatment differences and corresponding nonadjusted p-values were calculated. A 5% significance level (α = 0.05 or two-sided p < 0 0.05) was applied for the comparison of treatment groups. p-values represent the interaction between treatment and time. If significance was detected, a Bonferroni post hoc was applied. Model assumptions for the repeated measures mixed-effects model were also assessed. Residuals were visually inspected for normality and homoscedasticity using diagnostic plots. Point estimates, 80% CIs, and 95% CIs were extracted from the analysis of covariance model. All analyses were performed using SAS® version 9.4 with SAS Enterprise Guide version 8.3. For within-group analysis, a comparison of means from baseline was performed using a two-tailed Welch’s t test. Total area under the curve (tAUC) was calculated using the Time Response Analyser [35] to assess CK and CRP responses to exercise over time. Statistical analysis was conducted on raw data, with supporting descriptive statistics (% change) and effect sizes (Cohens d) included in text where appropriate. Effect sizes using Cohen’s d were calculated using the difference between group means divided by the pooled standard deviation, and interpreted using thresholds of > 0.2, > 0.5, and > 0.8 for small, moderate, and large effect sizes, respectively [36]. The pooled standard deviation was computed as the square root of the weighted average of the two groups’ variances, accounting for sample size. A formal power calculation was not done prior to commencing the study given the lack of relevant literature assessing supplements targeting mitochondrial function and subsequent impact on running performance related outcomes in well-trained individuals. Sample size was therefore estimated based on similar studies exploring mitochondrial targeting supplements and endurance performance [37, 38]. Figures were produced using GraphPad Prism (version 10.4.1, GraphPad Software Inc., La Jolla, CA, USA). The schematics in Figs. 1 and 4A were created using BioRender.com.
Comments (0)