PIMT represents the most common mesenchymal lung tumor in individuals under 16 years, yet accounts for less than 1% of all lung tumors [14]. Previous studies predominantly indicate no significant influence of gender on PIMT occurrence [15]. Three prior Korean studies reported male proportions of 67%, 82%, and 62.5%, respectively among PIMT patients [16,17,18]. Our study revealed a male proportion of 57.1%, a variation potentially attributable to sampling differences across case series. This study provides further supplementation to the epidemiological data for this disease. Some studies suggest an endobronchial location may be a relatively common site for PIMT [18]while a retrospective study including 8 PIMT patients showed 75% occurred in the right lung [19]. Similarly, our study found a higher incidence in the right lung (64.3%) compared to the left lung(35.7%), consistent with the aforementioned result.
The etiology and pathogenesis of PIMTs remain unclear. Notably, patient eight had pre-existing MIA and developed a new ipsilateral PIMT. Histopathological analysis excluded recurrence or metastasis, suggesting a second primary tumor. This may imply independent oncogenic pathways for adenocarcinoma and PIMT, warranting further genetic analysis. To our knowledge, this is the first report of PIMT developing after lung adenocarcinoma resection. To date, no direct evidence suggests an association between PIMT occurrence and previous pulmonary surgery. The 50-month disease-free survival (DFS) post PIMT resection in this case reinforces its distinct biological behavior.
The clinical symptoms of PIMT are non-specific, primarily including cough, hemoptysis, chest pain, and dyspnea. Approximately 30% of patients may present with systemic symptoms such as fever and weight loss [20, 21]. In our study, four patients (28.6%) exhibited cough as the predominant symptom, which may be associated with local tumor invasion and inflammation. Four patients were asymptomatic, and one case was incidentally discovered following chest trauma, further illustrating the non-specific nature of PIMT symptoms.
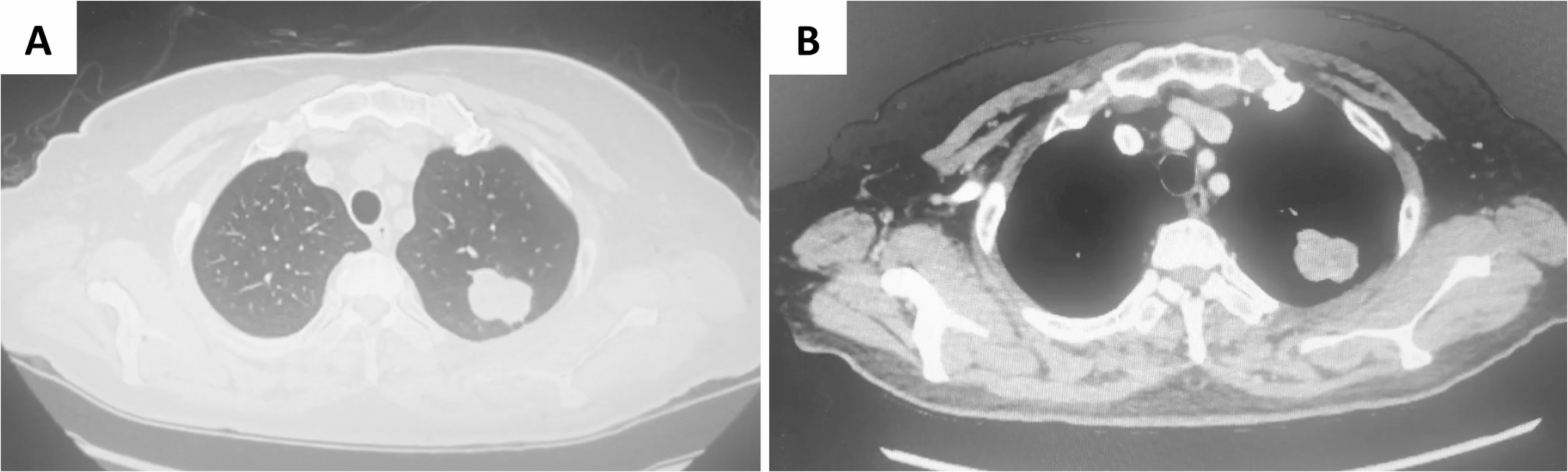
Preoperative definitive diagnosis of PIMT is challenging. Differentiating PIMT from lung cancer, carcinoid tumors, and specific pulmonary infections such as tuberculosis, aspergillosis, cryptococcosis, and hamartoma can be difficult [22]. Therefore, routine examinations are essential prior to pulmonary surgery. CT and PET-CT are commonly employed auxiliary examinations for PIMT, though their findings may also lack specificity [23]. CT is utilized in nearly all PIMT cases, typically revealing a solitary pulmonary nodule of variable morphology. Some may appear as round, high-density nodules without lymphadenopathy [24]. Contrast-enhanced CT predominantly demonstrates varying degrees of delayed enhancement. The density may appear heterogeneous or exhibit central low-density necrosis. Mild delayed enhancement may be observed upon contrast administration, potentially related to abundant tumor vascularity accompanied by partial inflammation [25].The role of FDG PET-CT in IMT is limited. A previous study including 7 IMT patients and one spindle cell sarcoma patient transformed from IMT reported a mean SUVmax of 10.9 ± 5.5 (range, 3.3 to 20.8) [26]. Its utility is confined to monitoring local recurrence, treatment response, and distant metastasis. Another study has found diagnostic value of PET-CT in IMT in cases associated with other systemic diseases, such as G-CSF-producing inflammatory myofibroblastic tumor [27]. In our series, 6 patients underwent preoperative 18F-FDG PET/CT, revealing variable FDG uptake ranging from low to high intensity, with a mean SUVmax of 9.6 ± 2.7 (range, 5.6 to 26.5). This variability may be related to tumor density, biological behavior, and the activation level of inflammatory cells. It is critical to note that high SUVmax values may mimic malignancy, necessitating pathological confirmation to avoid overtreatment.
Although rare cases of spontaneous regression [28] of PIMT or cure following short-term hormonal therapy [29], surgical resection remains the preferred approach for PIMT. Frozen section examination is crucial for establishing a diagnosis intraoperatively and determining the extent of resection, including the decision for lymph node dissection. However, intraoperative frozen section diagnosis of PIMT is also challenging [16, 30]. Yüksel C et al. [31] performed frozen section analysis on 10 surgical patients ultimately confirmed as PIMT by final pathology, only 2 patients (20%) were diagnosed as PIMT intraoperatively. In contrast, in our case series, frozen sections suggested PIMT in nine patients (64.3%), reflecting greater expertise among our pathologists in identifying these tumors. Considering reports of PIMT recurrence characteristics in some studies, our approach is to perform a moderately wider resection when feasible and tolerated by the patient. No lymph node metastasis occurred in 5 patients undergoing dissection, reinforcing that routine lymphadenectomy is unwarranted. We recommend selective dissection only when malignancy cannot be excluded intraoperatively, consistent with the findings of Thistlethwaite et al. [32]. Data regarding survival rates in PIMT are limited in the literature. Studies by Zhu (eight patients) [19], Jeong (eight patients) [18]and Lee(15 patients) [16] reported no mortalities, with a 100% overall survival (OS) rate, which aligns with our 100% OS outcome. Importantly, our cohort also demonstrates the longest documented follow-up (122 months) among surgically resected PIMT cases with 100% DFS. Nevertheless, some studies report that recurrence may occur even after surgery [11, 33]. Therefore, patients should undergo close surveillance following resection to detect local or distant recurrence. Notably, survival analysis was not performed in our study as all patients remained alive with no recurrence or metastasis.
The histological presentation of PIMT exhibits considerable heterogeneity. A relatively consistent feature is the presence of proliferating myofibroblasts accompanied by varying degrees of mononuclear inflammatory cell infiltration [22]. The inflammatory infiltrate is diverse, frequently composed of lymphocytes and plasma cells, with occasional eosinophils [34]. The density of this infiltrate may obscure the underlying myofibroblasts. Notably, the myofibroblast, the core cellular component of PIMT, represents a distinct cell type intermediate between smooth muscle cells and fibroblasts. Its unique characteristics often render definitive diagnosis challenging even on intraoperative frozen section examination, underscoring the critical importance of immunohistochemical staining for final confirmation [35]. Immunophenotypically, PIMT characteristically expresses mesenchymal markers. In our study, cells demonstrated strong immunoreactivity for SMA in 85.7% of cases, aligning with the general observation of SMA positivity in approximately 80–90% of cases in another reserach [36]. ALK protein expression is also positive in approximately 50–75% of cases [38]with ten cases (71.4%) of patients in our study showing ALK positivity. Additionally, some literature suggests that a Ki67 proliferation index exceeding 25% signifies substantially increased tumor aggressiveness [37]. Notably, all Ki67-positive patients exhibited a Ki67 index ≤ 15% (range, 5–15%) in our cohort. This low proliferative activity may contribute to the absence of recurrence in our cohort. However, histological classification appears to have limited prognostic value, as all patients after R0 resection achieved favorable outcomes regardless of their histopathological subtype.
When surgical resection is not feasible, alternative treatments including radiotherapy, chemotherapy and systemic corticosteroids—may serve as substitutes [34, 38]. ALK tyrosine kinase inhibitors (TKIs), represented by crizotinib, hold significant promise for patients with locally unresectable, advanced, or metastatic ALK-positive IMT [39]. Long-term follow-up indicates a median progression-free survival (PFS) of 18 months and the OS rate of 83.3% in ALK-rearranged patients treated with crizotinib [40]. Although these studies encompassed IMTs from all anatomical sites, the high prevalence of ALK gene rearrangements in PIMT patients similarly supports confidence in the potential benefit of ALK-positive TKIs for PIMT.
While immunotherapy has demonstrated substantial potential in treating numerous malignancies, reports on using immune checkpoint inhibitors for IMT patients are limited. In an analysis of 35 IMT samples by Cottrell et al. [41]28 samples (80%) exhibited PD-L1 positivity in immune cells. Among 20 recurrent and metastatic tumor samples, 16 (80%) were PD-L1 positive. Of eight ALK-negative tumor samples, seven(88%) showed PD-L1 positivity. However, it is critical to note that PD-L1 testing was not performed in our cohort since all patients underwent R0 resection and PD-L1 testing is not routinely indicated for resected PIMT. This PD-L1 positivity suggests that immunotherapy may have therapeutic potential in PIMT. Nevertheless, given the limited availability of clinical reports and the lack of approved indications, it is imperative to conduct prospective trials in the future to validate the effect of PD-L1 positivity for PIMT, especially for those of ALK-negative or unresectable patients.
Though the significance of our study lies in providing crucial clinical insights and favorable prognostic data for this rare disease, the key limitations include its retrospective single-center design and small sample size, inherent to rare disease research. The absence of pediatric cases limits generalizability. Therefore, additional clinical data are required to identify clinical and prognostic features of PIMT.
To the best of our knowledge, this study presents one of the largest single-center surgical series of PIMT cured by surgery; however, our understanding of PIMT remains significantly limited. Due to the lack of specific diagnostic tools, current diagnosis relies on pathology. Surgical resection remains the preferred and effective treatment modality for PIMT, typically yielding satisfactory outcomes. Targeted therapy demonstrates considerable promise, particularly for advanced and ALK-positive PIMT. Given the risk of recurrence and metastasis in some cases, we recommend close, long-term follow-up. Future studies with larger cohorts are warranted to enhance our understanding of PIMT.



Comments (0)