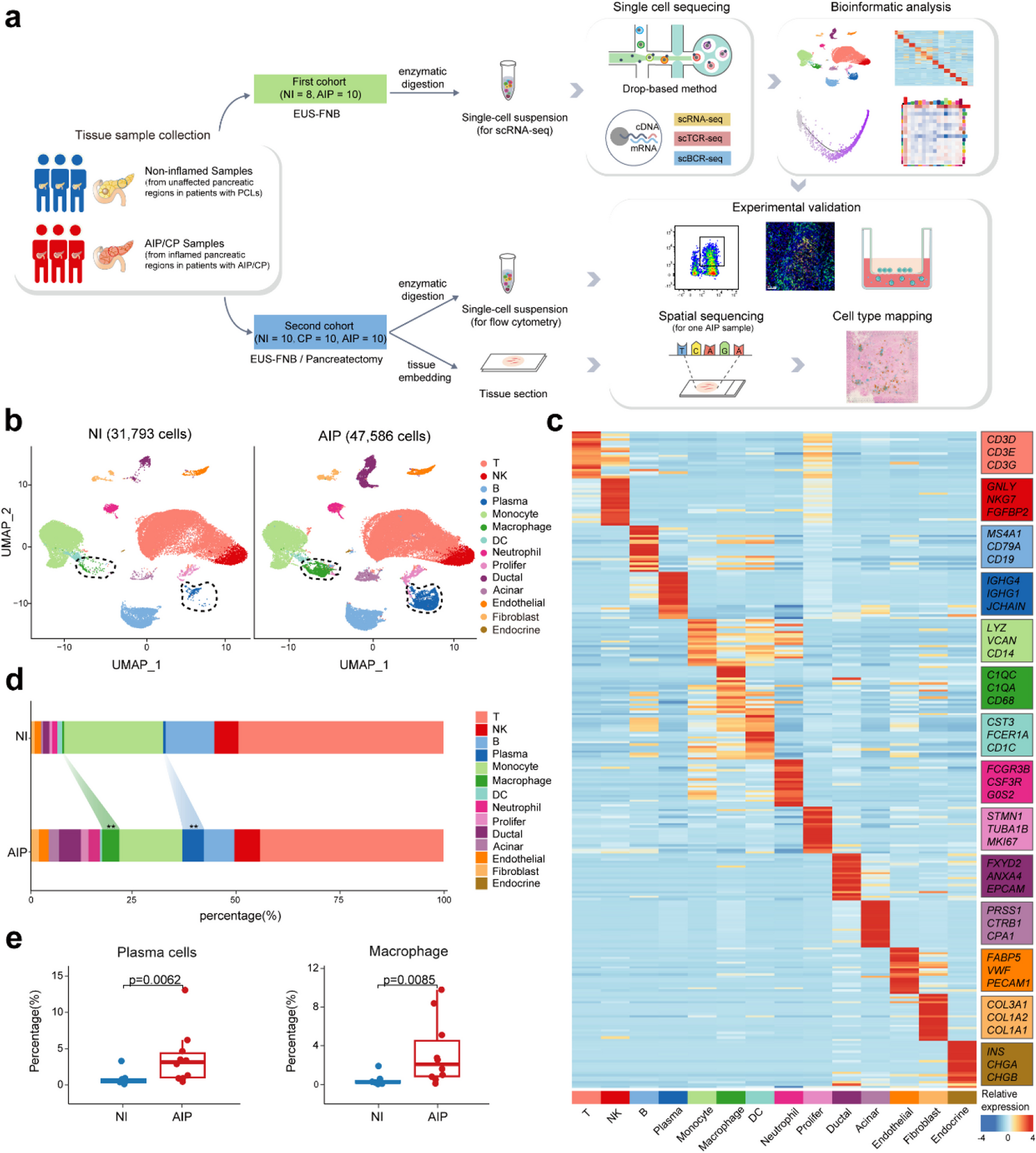
Human specimens
Human pancreatic tissues used for scRNA-seq were collected from two groups via endoscopic ultrasound-guided fine-needle aspiration/biopsy (EUS-FNA/B) from Ruijin Hospital: specifically, the inflamed pancreatic regions of 10 patients with AIP, and the adjacent normal non-inflamed pancreatic tissues of 8 patients with pancreatic cystic lesions (PCLs). All enrolled AIP patients had diffuse pancreatic lesions and required histopathological evidence via EUS-FNA/B to establish a definitive diagnosis, in accordance with the International Consensus Diagnostic Criteria (ICDC) for type 1 AIP [1]. During the sample collection procedure, an experienced endoscopist performed endoscopic ultrasound (EUS) after administering intravenous anaesthesia. A 22G Franseen needle (Acquire, Boston Scientific, Natick, MA) was used for sampling, with the puncture site selected based on the presence or absence of jaundice. Once the rapid on-site evaluation confirmed sample quality, an additional puncture was conducted for research purposes. For patients with PCLs, a 22G Expect needle (Boston Scientific, Natick, MA) was employed. The initial puncture was used to obtain adjacent normal pancreatic tissue, while a subsequent puncture was performed to collect cystic fluid for clinical diagnosis. An additional cohort comprising 5 AIP patients and 5 PCLs patients were enrolled and we collected their pancreatic tissues for flow cytometry following the same procedure described above. From these participants, we also collected their peripheral blood samples for chemotaxis assay and enzyme-linked immunosorbent assay (ELISA).
Pancreatic samples for immunofluorescence or ST were collected from 5 AIP patients, 10 CP patients, and 5 PCLs patients who underwent total pancreatectomy from Ruijin Hospital. The initial suspicion was pancreatic ductal adenocarcinoma (PDAC) before surgery; however, AIP or CP was confirmed by pathology following the procedure. Their peripheral blood samples were also collected for ELISA.
All biopsies and surgeries occurred before a definitive diagnosis was established, ensuring that all participants were treatment-naive at the time of sampling. All procedures for sample collection adhered to standard clinical practices. The demographics and characteristics of control and AIP patients included in the scRNA-seq analysis (cohort 1) are detailed in Table 1 and Additional file 1: Table S1. For the validation experiments (cohorts 2 and 3), demographic information for control, CP, and AIP patients is provided in Tables 2 and 3 and Additional file 1: Table S2-S3.
Table 1 Statistical comparisons of demographic data and patient characteristics between non-inflamed control (NI) group and AIP patients for scRNA-seq analysis (cohort1)Table 2 Statistical comparisons of demographic data and patient characteristics among NI group and AIP patients for flow cytometry experiments (cohort2)Table 3 Statistical comparisons of demographic data and patient characteristics among NI group, CP and AIP patients for immunofluorescence or ST experiments (cohort 3)Tissue processing and single-cell isolation
Fresh pancreatic tissues were preserved in 2x protease inhibitor (Solarbio, P6730) mixed with complete RPMI 1640 medium (containing 10% foetal bovine serum (FBS), 100 U/mL penicillin, and 100 mg/mL streptomycin) on ice for transport. Tissue processing began within 30 min of tissue acquisition after EUS-FNA/B. Upon arrival, the tissues were washed with ice-cold phosphate-buffered saline (PBS) and cut into 2-mm pieces. Each sample was then enzymatically digested using a solution of 1 mg/mL Trypsin inhibitor (Sigma, T6522), 0.82 mg/mL Dispase (Sigma, D4693), 1 mg/mL collagenase VIII, and 0.15 mg/mL DNase I in 4 mL complete RPMI 1640 medium. Digestion was stopped by adding 4 mL of complete RPMI 1640 medium. Red blood cells were lysed with 2 mL ACK lysing buffer (Thermofisher, A1049201) for 5 min on ice. After two washes with 1 × PBS (Corning), the cell pellets were resuspended in single-cell buffer. The cell suspensions were filtered through a 70-μm cell strainer (BD) and 1% protease inhibitor was added. The freshly prepared single-cell suspensions were then used for scRNA-seq and flow cytometry.
Flow cytometry
Fresh cells from AIP pancreatic tissues and non-inflamed tissues were washed and incubated with Live/Dead dye (BV510, Biolegend, 1:1000) in PBS for 10 min at 4 °C. After incubation, cells were washed in PBS with 2% FBS and 2 mM ethylenediaminetetraacetic acid (EDTA), and this solution was defined as fluorescence-activated cell sorting (FACS) buffer. To minimize non-specific protein binding, myeloid cells were treated with human Fc Block (1:100) at 4 ℃ for 20 min. Surface markers were then stained with a panel of antibodies diluted in FACS buffer at the specified concentrations. The staining was conducted at 4 °C for 30 min using the following antibodies: Anti-CD45 (BD Horizon, 563792, 1:200), anti-CD4 (Biolegend, 317434, 1:200), anti-CD19 (Biolegend, 363024, 1:200), anti-CD3(BD Horizon, 563725, 1:200); anti-CD11c (Biolegend, 301644, 1:50), anti-IgD (Biolegend, 348240, 1:100), anti-PD-1 (Biolegend, 329910, 1:200), anti-CD38 (Biolegend, 303524, 1:200), anti-CD27 (Biolegend, 302815, 1:200). After staining, the cells were washed twice. Cells were then fixed (Fixation Buffer, BD), permeabilized (Intracellular Staining Permeabilization Wash Buffer (10X), BD) and subjected to intracellular labelling using anti-CD68 (Biolegend, 333806, 1:200). Flow cytometry was performed on a BD Symphony (BD Biosciences) with data acquisition managed by BD FACSDiva software v8.0.2 and analysis performed using FlowJo v.10.81 (Tree Star Inc.). Statistical comparisons were carried out using a paired two-tailed t-test with GraphPad Prism 6.
Multiplex immunofluorescent (mIF) staining
Human pancreatic tissue samples were fixed in 4% paraformaldehyde for 24 h, followed by dehydration and paraffin embedding. They were then sectioned into 5-μm slices for use. Multiplex immunofluorescence was performed using PanoPANEL Kits (Panovue, 10234100050) according to the manufacturer’s instructions. The procedure began with deparaffinization of the slides using xylene and a series of ethanol washes (100%, 95%, and 70%). This was followed by microwave-based antigen retrieval and a 30-min antibody blocking step. Primary antibodies were incubated at room temperature for 1 h, followed by incubation with HRP-labelled secondary antibodies for 30 min at room temperature. The slides were then treated with Tyramide signal amplification (TSA) fluorescent dye working solution for 30 min. The following antibodies and corresponding fluorescent dyes were used for mIF staining: panel1: CD19 (Abcam, ab134114, 1:500, PPD480); CD11c (Abcam, ab52632, 1:500, PPD520); CD4 (Abcam, ab133616, 1:500, PPD570); PD1 (Abcam, ab52587, 1:100, PPD650); CD23 (Abcam, ab16702, 1:100, PPD780); panel2: CD68(Abcam, ab955,1:1000, PPD520); CXCL9 (Abcam, ab290643, 1:100, PPD650). After staining, nuclei were counterstained with DAPI for 20 min, and the slides were then sealed with Nail Polish. Scanning of the slides was performed using the SLIDEVIEW VS200 (Olympus).
HALO spatial analysis
Images from mIF were analysed with HALO software (Indica Labs, Corrales, NM, USA). Quantification of cells and staining was performed according to HALO HighPlex FL analysis instructions. The Tissue Classifier function was employed to distinguish between tertiary lymphoid structures (TLSs) and non-TLS areas using the Random Forest algorithm, as illustrated in Fig. 5d. And the infiltration of age-associated B cells (ABCs) and Tfhs to peripheral boundary of TLS was evaluated by using infiltration analysis related to Fig. 5e. Proximity analysis was conducted to evaluate (1) the percentage of ABCs (CD19+CD11c+) within 100 μm (divided into 10 equal ranges) of Tfhs (PD1+CD4+) related to Fig. 5f and (2) the percentage of ABCs located within or outside a 50-µm radius of Tfhs, as depicted in Fig. 5g.
B cell subset isolation
B cells were isolated from the peripheral blood of AIP patients or healthy people using MojoSort™ Human B Cell (CD43−) Isolation Kit (Biolegend, 480061) according to the manufacturer’s instructions. The purity of isolated cells was verified to be more than 95%. The collected cells were used for functional chemotaxis and co-culture experiments.
Macrophages culture
THP-1 cells (Quicell, Quicell-T239) were plated at 1 × 105 per well of 24-well plates and cultured in RPMI-1640 supplemented with 10% FBS, 1% penicillin/streptomycin, and 50 mM β-mercaptoethanol at 37 °C in a humidified atmosphere of 5% CO2. THP-1 derived M1-type macrophages were obtained by culturing THP-1 cells with 100 ng/ml phorbol ester (PMA) (Beyotime, S1819) for 24 h and subsequently stimulated with 100 ng/ml lipopolysaccharides (LPS) (MCE, HY-D1056) for 24 h.
Transfection
THP-1 derived M1-type macrophages were cultured to reach the confluency of 70% before the transfections. Negative control or CXCL9-targeting small interfering RNA (siRNA) were prepared and mixed with lipofectamine RNAiMAX (Invitrogen, 13778150) according to the manufacturer’s instruction. After the addition of siRNA-lipofectamine RNAiMAX, cells were incubated for a further 24 h before chemotaxis experiments.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from THP-1 derived M1-type macrophages transfected with either si-negative control or si-CXCL9 using Trizol (Invitrogen). cDNA was then synthesized from SuperScript IIIcDNA Synthesis Kit (Invitrogen). mRNA expressions were detected with SYBR Green on a 384 well real-time PCR system (Applied Biosystems, QuantStudio 6 Flex). Primer sequences were listed as follows: CXCL9, Antisense 5′—CCAGTAGTGAGAAAGGGTCGC – 3′, and Sense5′- AGGGCTTGGGGCAAATTGTT −3′; GAPDH, Antisense5′- ACAACTTTGGTATCGTGGAAGG −3′, and Sense 5′- GCCATCACGCCACAGTTTC −3′; siCXCL9, Antisense 5’—UUUCUCACUACUGGGGUUCTT—3′, and Sense 5′- GAACCCCAGUAGUGAGAAATT—3′. We utilized 2−ΔΔCt method to calculate the normalized expression of each gene relative to gene GAPDH.
Chemotaxis assay
Isolated B cells from the peripheral blood of AIP patients were resuspended at 1 × 106 cells/ml in preheated RPMI1640 supplemented with 0.5% BSA. For anti-CXCR3 group, the B cells were incubated for 30 min at 37 °C with anti-CXCR3 blocking antibody (Biolegend, 353702, 1:100). Then, in the lower chamber of the Transwell apparatus (Corning, Transwell-24 well with 5-µm pore polycarbonate membrane, #3421), 600 µl of recombinant human CXCL9 (Peprotech, 300-26-5UG) was added to achieve a final concentration of 100 ng/ml. In the upper chamber, 200 µl of cell suspension was added. Wells with added vehicle served as negative controls. For macrophages-B cell chemotaxis assay, Transwell apparatus (Corning, Transwell-24 well with 5-µm pore polycarbonate membrane, #3421) were inserted into the 24-well plates with the si-negative control or si-CXCL9 transfected macrophages cultured in. Two hundred microlitres of B cell suspension was added into the upper chamber. After incubating at 37 °C for 3 h, migrating cells were collected from the lower chamber and evaluated by flow cytometry according to the protocol described above. All assays were performed in triplicate.
T cell subsets isolation and co-culture experiment
T cells were isolated from the peripheral blood of AIP patients using FACS Arial III (BD Bioscience) and sorted as PD1+CD4+Tfhs and PD1−CD4+T helper cells (Ths). The collected cells were used for functional co-culture experiments. Sorted Th and Tfhs were plated at 1 × 104 cells per well of 96-well round-bottom plate and cultured in RPMI-1640 supplemented with 10% FBS and 1% penicillin/streptomycin. They were then stimulated with CD3/CD28 beads (Easylso, AH2001) or 50 ng/ml IL-21R-Fc (UA BIOSCIENCE, UA011187) for 48 h and further co-cultured with isolated healthy blood B cells at a ratio of 1:5 in the final volume of 200 µL for 6 days. Cells were collected and evaluated by flow cytometry according to the protocol described above.
ELISA detection
The peripheral blood of AIP, CP, and control group patients was centrifuged at 1800 rpm (757 g) and room temperature for 5 min. The plasma was obtained for the ELISA detection. Fifty microlitres of plasma from each sample was transferred to a 96-well plate for the detection of CXCL9 using Human CXCL9 ELISA Kit (ml060599, Mlbio) following the manufacturer’s protocol. Finally, the absorbance at 450 nm was measured using a Multiskan Go (Thermofisher).
Single-cell RNA-seq, TCR/BCR-seq, and data processing
Single-cell suspensions were prepared according to the manufacturer’s protocol for the 10x Chromium Next GEM Single Cell 5’ Kit v2 (10x Genomics, PN-1000263) to generate libraries for single-cell transcriptome and TCR/BCR V(D)J analysis. Sequencing was then conducted on an Illumina NovaSeq 6000 platform at the Shanghai Institute of Immunology. Sequence processing was performed using the standard Cell Ranger pipeline (10x Genomics), with data mapped to the GRCh38 reference genome using Cell Ranger Software (v5.0.1). The resulting preliminary count matrices were analysed with Seurat (v4.3.0) [16]. Genes expressed in fewer than three cells were removed, and cells with over 10% mitochondrial genes, 25% red cell genes, fewer than 200 or more than 7000 gene counts, or UMI counts outside the range of 500 to 50,000 were filtered out. Potential doublets were identified and removed using Scrublet (v0.2.3) and DoubletDetection (v3.0) [17, 18]. The data was then normalized and transformed to a log scale using the “NormalizeData” function.
Spatial transcriptomics sequencing and data processing
Formalin-fixed paraffin-embedded (FFPE) 5-μm pancreatic lesion tissue sections from a representative AIP patient were used. Haematoxylin and eosin (H&E) staining was performed on the sections firstly. The sections were then de-crosslinked and hybridized with transcriptomic probes to detect the presence of target RNA. After probe hybridization, the Visium CytAssist system was used to transfer these transcriptomic probes from the glass slides to the Capture Area on the Visium slide. Following this, library preparation and sequencing were carried out using the 10x Visium CytAssist Spatial Gene Expression for FFPE, Human Transcriptome, 6.5 mm kit (10x Genomics, PN-1000520) according to the manufacturer’s protocol. Sequencing was performed on an Illumina NovaSeq 6000 platform at Xu Ran Biological (Shanghai, China). Raw sequencing reads from the ST data were quality-checked and mapped to the GRCh38 reference genome for human samples using Space Ranger (v2.1.0).
Dimension reduction and unsupervised clustering
The top 4000 most variable genes were identified using the “FindVariableFeatures” function. Gene expression levels were then scaled by regressing out unwanted sources of variation, including total counts, mitochondrial gene counts, and ribosomal gene counts. These variable genes were used for principal component analysis (PCA), and the top 20 principal components were selected for subsequent analyses. Graph-based clustering was performed using the “FindNeighbors” and “FindClusters” functions to identify cell subtypes. Dimensionality reduction was achieved using Uniform Manifold Approximation and Projection (UMAP). For unsupervised clustering, cells were initially categorized into major types, including T cells, NK cells, B cells, plasma cells, myeloid cells (monocytes, macrophages, dendritic cells, and neutrophils), acinar cells, ductal cells, proliferating cells, fibroblasts, endothelial cells, and endocrine cells, based on canonical cell markers. Subsequently, specific subtypes within B cells, CD4+ T cells, CD8+ T cells, and macrophages were further resolved at higher resolutions. Any unexpected gastric and duodenal epithelial cells introduced during the EUS-FNA/B sampling procedure were removed from the analysis.
Analysis of differentially expressed genes
Differentially expressed genes (DEGs) between clusters were identified using the “FindAllMarkers” function with a minimum percentage threshold of 0.1 and a log fold-change threshold of 0.25. DEGs were visualized using a heatmap after log transformation and scaling of gene expression data. The Wilcoxon rank-sum test Find Clusters was applied to calculate p-values for the comparisons, and p-values were adjusted for multiple testing using the Bonferroni correction. A comprehensive list of canonical and signature marker genes for each cell cluster is provided in Additional file 1: Table S4-8.
Functional annotation of genes and Gene Set Enrichment Analysis (GSEA) analysis
Gene Ontology biological process (GO BP) analysis was performed using the clusterProfiler package (v4.0.5) [19] to conduct functional enrichment analyses of DEGs. The analysis utilized a p-value to assess the probability of observing pathway members, based on an accumulative hypergeometric distribution. TLS scores were calculated using the ing the f observing pathway members, based on an accumulative hypergeometric distribution. nature marker genes for each cell cluste [20] and are listed in Additional file 1: Table S9. Additionally, gene sets from the C5: ontology gene set collection in The Molecular Signatures Database (MSigDB, http://software.broadinstitute.org/MSigDB/msigdb/) [21] were downloaded. GSEA was performed using the GSEA function in the clusterProfiler package (v4.0.5) [19] to identify significantly differentially enriched annotated gene sets between macrophages from the AIP group and those from the non-inflamed group. Gene signatures or pathways with a false discovery rate (FDR) less than 0.05 were considered significantly enriched.
Trajectory analyses
Developmental trajectory analyses of ABCs and plasma G cells were realized with Monocle2 (v2.22.0) [22]. The top 2000 highly variable genes in these cells were selected as input for the analysis. Dimensionality reduction was performed using the “DDRTree” method. Differentially expressed genes along the pseudotime trajectory were identified with the “differentialGeneTest” function, applying a q-value threshold of < 0.01. The results were visualized using the “plot_pseudotime_heatmap” function.
Cell–cell interaction analysis
Intercellular communication of ligand-receptor pairs was analysed using CellChat (v1.1.3) [23]. The CellChatDB.human database was utilized for signalling pathways. The communication probability between interacting cell clusters was calculated using the “computeCommunProb” function. The “netVisual_x” functions were employed to visualize the strength of cell–cell communication networks and significant ligand-receptor pairs. Additionally, NicheNet (v2.0.0) [24] was used to infer crosstalk between CD4+ T cells and IgD− ABCs. For ligand-receptor interactions, genes expressed in more than 10% of the cells within each cluster were considered. CD4+ T cells were designated as sender cells, while IgD− ABCs were considered as affected cells, with other B cell subsets used as reference cells. The top 10 ligands from the sender cells and the top 250 targets of differentially expressed genes in the affected cells were selected for paired ligand-receptor activity analysis. The regulatory activity of ligands was visualized using the “ligand_activity_target_heatmap” function in the NicheNet output.
Single-cell TCR/BCR-seq analysis
TCR and BCR clonotype assignment was performed using the Cell Ranger (v5.0.1, GRCh38 reference genome) VDJ pipeline (10x Genomics) to obtain diversity metrics, including clonotype frequency and barcode information. Subsequent analysis was conducted using the scRepertoire package (v2.0.0) [25]. For BCR analysis, each cell was assigned the pair of heavy and light chains with the highest UMI counts. The strict definition of a clone is based on the normalized Levenshtein edit distance of CDR3 nucleotide sequences and V-gene usage via the function “combineBCR”. For TCR analysis, each cell was assigned the pair of alpha and beta chains with the highest UMI counts. Unique CDR3 amino acid sequences were defined as clones, and cells with identical clones were considered to have originated from the same ancestor. TCR/BCR clones were correlated with scRNA-seq data and projected onto UMAP plots based on barcode information. To analyse the origin of plasma cells, the assigned the pair of alpha and beta chains with the highest UMI counts. Unique CDR3 amino acid sequences were defined as clones, and cells with identical clones function. The “ClonalDiversity” function assessed TCR/BCR clonotype diversity using metrics such as Shannon entropy, inverse Simpson index, normalized entropy, Gini-Simpson index, Chao1, and ACE. Additionally, the “clonalOccupy” and “clonalOverlap” functions were used to evaluate the clonal size and overlap of each T cell or B cell subpopulation, respectively.
Spatial deconvolution and annotation in ST
To determine the cell composition of spots on spatial transcriptomics slides, we utilized the Cell2location Python package (v0.1.3) [26]. The analysis began with preprocessing the raw data, including standard quality control and filtering to exclude low-quality spots and genes with minimal expression. The data was then normalized using the total-count normalization method provided by Cell2location. We trained the Cell2location model using our integrated scRNA-seq atlas alongside the ST data. This model predicted the expression of each gene for each cell type at each spot in the tissue, enabling us to identify and visualize the spatial distribution of CXCL9+ macrophages, IgD− ABCs and Tfhs within the tissue. Additionally, we used the ding standard quality control and filtering to exclude low-quality spots and genscore.
Tissue distribution of immune cell subsets among AIP, CP, and NI groups
ScRNA-seq data of CP were acquired from the Gene Expression Omnibus (GEO) database at https://www.ncbi.nlm.nih.gov/geo/ in GSE165045 [27]. To eliminate the batch effect, we performed harmony algorithm in R package Harmony v0.1.0 [28] to remove batch effect before integrating analysis. To compare the immune microenvironment among AIP, CP, and control groups, we utilized the “FindTransferAnchors” and “TransferData” functions in Seurat. This approach involved evaluating the cell composition of the pancreas from CP patients by using scRNA-seq data from CP as the query set and our dataset as the reference. We calculated the ratio of observed to expected cell numbers (Ro/e) for each cell subset across different tissues (AIP, CP, and non-inflamed) to quantify the preference of each subset. The expected cell numbers for each cell subset and tissue combination were derived from the χ2 test. A cell subset was considered enriched in a specific tissue if RO/E was greater than 1.
Statistical analysis
Statistical analyses were conducted using R or GraphPad Prism 6, with a significance level set at P < 0.05. The methods used included two-tailed t-tests, Wilcoxon rank-sum tests, and one-way analysis of variance (ANOVA), as specified in the figure legends. For multiple comparisons, Bonferroni correction was applied.









Comments (0)