Tissue collection
106 pairs of HCC and corresponding adjacent normal liver tissues were obtained from patients with HCC who underwent hepatectomy at Zhongshan Hospital of Xiamen University from 2013 to 2022. Prior to the surgery, these patients did not undergo interventional therapy, chemotherapy, or targeted therapy. Tissue samples were collected, immediately fixed in 10% neutral formalin solution. Written informed consent for this research was obtained from all patients. The informed consent document and experimental procedures were approved by the Ethics Committee of Zhongshan Hospital of Xiamen University in accordance with the World Medical Association Declaration of Helsinki.
Cell culture
HEK293T, Huh7, and SK-Hep-1 cells were purchased from the Cellcook Company (Guangzhou, China). The authentication of these cell lines was performed through comparison with the STR database, and all experiments were performed with mycoplasma-free cells. The cells were cultured in high-glucose Dulbecco’s modified Eagle medium (DMEM; Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Vivacell, Shanghai, China) as well as Penicillin-Streptomycin (Gibco, USA) at 37 °C in a humidified incubator with 5% CO2 in the air.
Immunohistochemical (IHC) staining
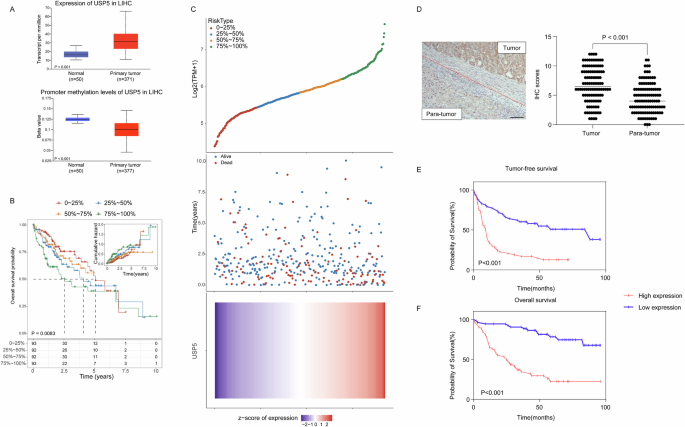
Fixed tissues were processed using the Tissue-Tek TEC5 Embedding Centre (Sakura Finetek Co., Japan), following a standard protocol, and embedded in paraffin blocks as 4-μm thick sections. Briefly, IHC staining proceeded as follows: sections were deparaffinized, hydrated, and immersed in 3% H2O2 for 20 min at room temperature. Subsequently, they were incubated overnight at 4 °C with anti-USP5 (1:10000, ab10800, Abcam) and anti-FASN (1:1000, 10800-1-AP, Proteintech) antibodies. Slides were then treated with biotinylated goat anti-rabbit antibodies for 1 h and stained with diaminobenzidine (DAB; Maixin Biotechnology, China), followed by hematoxylin counterstaining (Maixin Biotechnology, China). To prevent bias, two clinical pathologists, blinded to experimental data, independently assessed IHC-stained sections. In summary, 100 cells were randomly counted in 200× microscopic fields and categorized into five groups based on the percentage of positive staining cells in HCC tissues: 0=negative; 1–3 = 1–25%; 4–6 = 26–50%; 7–9 = 51–75%; 10–12 = ≥ 76%. Scores from 0 to 6 indicated low-expression, while scores from 7 to 12 indicated high-expression.
Overexpression of USP5
Lentiviral particles expressing an empty vector and USP5-Flag were purchased from Public Protein/Plasmid Library (Jiangsu Province, China). The cells were infected with these lentiviruses in the presence of polybrene (8 μg/mL). After 48 h, the cells were used for the following experiments.
Knockdown of USP5 expression
Two different shRNAs targeting USP5 (shUSP5-1: CCACACGATTTGCCTCATT, shUSP5-2: TAGACATGAACCAGCGGAT) and one shRNA targeting FASN (shFASN: TACTGGATGCGTTCTTCAA) were inserted into pLV-shRNA-puro lentiviral, respectively. Lentiviral particles were produced in HEK293T cells according to the standard procedure. The lentiviral particles were collected 48 h after transfection. Cells were infected with viral supernatants using polybrene (8 μg/mL). After 48 h of infection, stable cells were selected using 5 μg/mL puromycin (Inovogen, China) for one week.
Cell proliferation detection
Cells were plated in triplicate at a density of 1 ~ 2 × 103 cells per well in 96-well culture plates. The cell proliferation was evaluated in three separate experiments through the use of the CCK-8 (Dojindo, China) assay at specified time intervals, following the manufacturer’s guidelines. The absorbance was quantified at 450 nm using a microplate reader (Infinite® F50, Tecan).
Transwell assay
To assess cell migration, the specified cells were resuspended with 100 μL serum-free DMEM and then applied to the upper chamber (8 μm pore size, Corning, USA). Cell invasion was tested using Matrigel Invasion Chamber (8 μm pore size, Corning, USA). The lower chamber was filled with 600 μL DMEM containing 10% FBS. Subsequent to 24 h of incubation, the cells that migrated or invaded were fixed with 4% paraformaldehyde at room temperature for 20 min, followed by staining with 1% crystal violet for 30 min. The cells were subsequently counted under a microscope.
Mass spectrometry
Huh7 cells were treated with 10 μM MG132 (MedChemExpress) for 6 h. Cell lysates were subjected to immunoprecipitation using anti-USP5 antibodies (Cell Signaling Technology, USA) overnight. Subsequently, these immunoprecipitated complexes were combined with Dynabeads Protein G (Invitrogen, USA) for an additional 2 h incubation. Gel electrophoresis was performed, followed by in-gel digestion to prepare samples for subsequent proteomic analysis. The LC-MS/MS analysis aimed at identifying proteins interacting with USP5 was carried out by Applied Protein Technology (Shanghai, China).
Protein extraction and western blotting
Cell lysis was performed using radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime, China). Following quantification with a BCA protein assay kit (Thermo), protein samples underwent separation through sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequent transfer onto polyvinylidene difluoride (PVDF) membranes (Millipore, USA). After blocking with 5% non-fat milk (LABLEAD, China) for 1 h at room temperature, membranes were incubated with anti-USP5 (Cell Signaling Technology, USA), anti-β-actin (Axel Biotechnology, USA), or anti-FASN (Abcam, UK) antibodies. This was followed by incubation with anti-rabbit or anti-mouse secondary antibodies conjugated to horseradish peroxidase (HRP, Jackson). Immunoreactive proteins were then visualized using the enhanced chemiluminescence (ECL) detection system (Millipore, USA).
Co-immunoprecipitation
Total cellular proteins were extracted using co-immunoprecipitation (co-IP) lysis buffer (Beyotime, China). Cell lysates were incubated with anti-USP5 (Cell Signaling Technology, USA) or anti-FASN antibody (Abcam, UK) or a negative control IgG (Cell Signaling Technology, USA) and Dynabeads Protein G for immunoprecipitation (Invitrogen, USA) overnight at 4 °C. Subsequently, the beads were washed three times with lysis buffer, and the immunoprecipitated samples were collected for western blot analysis.
Animal study
A xenograft mouse model was established using 4-week-old male BALB/c athymic nude mice. A total of 2 × 106 Huh7 cells with shNC or shUSP5-1 or shUSP5-1 + FASN were subcutaneously injected into the nude mice, respectively. After approximately ten days, tumor size was assessed using a vernier caliper, and tumor volumes were calculated with the formula V = ½ (length × width2). 30 days post-injection, the mice were sacrificed under anesthesia. Then, the tumors were fixed in 10% neutral formalin, paraffin-embedded, and sectioned into 3 μm thick slices, which were used for further detection. All animal experiments were undertaken in accordance with relevant guidelines and granted approval by the Animal Care and Use Committee of Xiamen University.
Oil red O and Nile red staining
Cells were cultured in 6-well plates. At 70–80% confluence, a palmitic acid (PA; 0.25 mM; Kunchuang Biotechnology, Xi’an, China) and oleic acid (OA; 0.5 mM; Kunchuang Biotechnology, Xi’an, China) mixture in 0.5% BSA was added to the medium for 24 h. Cells were washed 3–4 times with PBS, fixed with 4% paraformaldehyde for 1 h, and incubated with Oil-Red O (Sigma, USA) for 1 h. Finally, the lipid droplets were visualized using light microscopy. For Nile red staining, cells were fixed with 4% paraformaldehyde solution, washed with PBS, and stained with Nile Red solution (7385-67-3, Solarbio, China) in the dark. In fluorescence microscopy, images were acquired after the samples were washed twice with PBS and stained with DAPI.
Targeted metabolomics
The lipidomic profiling was analyzed by Applied Protein Technology Company (Shanghai, China). In brief, tumor cells cultured to the logarithmic phase were treated with trypsin, centrifuged, and counted. The cells were then resuspended in the culture medium, adjusted to the desired concentration, plated in a 10 cm culture dish, and incubated overnight at 37 °C in a 5% CO2 atmosphere. When the cells reached 80% confluence, they were harvested and cryopreserved in liquid nitrogen. Samples were thawed on ice, and 50 µL of each sample was added into a 2 mL glass centrifuge tube, then mixed with 5 mL methylene dichloride-methanol (2:1 v/v). The mixture was incubated in an 80 °C water bath for 30 min for fatty-acid esterification. Then, 200 µL internal standard, 1 mL n-hexane, and 5 mL water were added and vortex mixed. The supernatant (500 µL) was subjected to GC-MS using an Agilent Model 7890–5977 system. To quantify medium- and long-chain fatty acids, a calibration curve for the concentration range of 0.1–2500 µg/mL was constructed. The internal standard was used to correct for injection variability and instrument response changes. Samples were separated with an Agilent DB-23 capillary GC column (60 m × 250 µm ID × 0.15 µm). The initial temperature was 50 °C for 3 min, increased to 220 °C at 10 °C/min, then to 250 °C at 15 °C/min, and held at 250 °C for 10 min. The carrier gas was helium (1.0 mL/min). Injection port and transmission line temperatures were 280 °C and 250 °C, respectively, under SIM mode.
Quantitative analysis of ubiquitination modifications
The ubiquitination proteomics was performed by Novogene (Beijing, China). Total Protein Extraction: Samples were ground in liquid nitrogen, lysed with 100 mM NH4HCO3 (pH 8), 8 M Urea, 0.2% SDS, and ultrasonicated on ice. Lysates were centrifuged (12000 g, 15 min, 4 °C) and the supernatant was collected. Proteins were reduced with 10 mM DTT (1 h, 56 °C) and alkylated with iodoacetamide (1 h, room temperature, dark). Samples were mixed with 4x volume of cold acetone and incubated at −20 °C for 2 h, then centrifuged. The pellet was washed with cold acetone and dissolved in 0.1 M TEAB (pH 8.5), 6 M urea. Protein Quality: Protein concentration was determined using the Bradford assay. 20 μg protein samples were analyzed on 12% SDS-PAGE, stained with Coomassie blue, and decolored until bands were clear. Peptide Preparation: 10 mg protein was digested with trypsin in 50 mM TEAB buffer (37°C, 4 h), followed by overnight digestion with CaCl2. pH was adjusted to <3 with formic acid, centrifuged, and the supernatant was desalted on a C18 column. Ubiquitin Peptide Enrichment: Lyophilized samples were dissolved in MOPS IAP buffer, mixed with anti-Ubiquitin Remnant Motif beads (2 h, 4 °C), washed, and eluted. Eluates were desalted and lyophilized. LC-MS/MS Analysis: Peptides were separated on a C18 column and analyzed with an EASY-nLCTM 1200 UHPLC system coupled to a Q Exactive HF-X mass spectrometer. MS and MS/MS settings included a full scan range of m/z 350–1500, resolution of 120,000 (MS) and 15,000 (MS/MS), AGC target values, and specific collision energies.
Statistical analysis
All statistical analyses were conducted utilizing SPSS software (Version 19.0). The data is presented as mean ± standard deviation (SD), and three independent experiments were carried out. Group-wise differences in the results were assessed using either Student’s t-test or a multi-way analysis of variance (ANOVA) test. The association between USP5 expression and clinical features of HCC patients was evaluated through the Chi-squared test. Spearman’s rank correlation was employed to scrutinize the relationship between USP5 and FASN expression. The Kaplan-Meier survival test and log-rank test were utilized to investigate the correlation between USP5 expression and the prognosis of HCC patients. Significance was assigned for p-values less than 0.05.

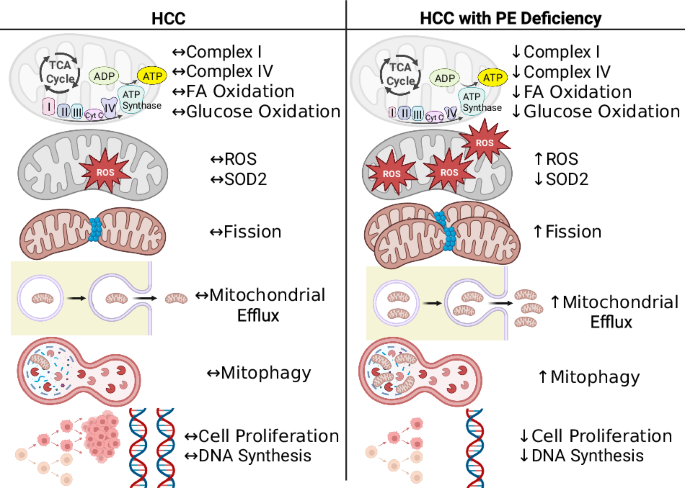
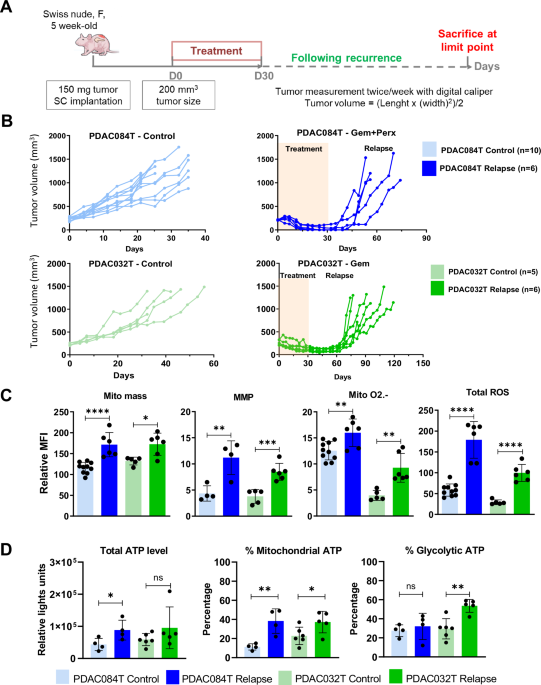
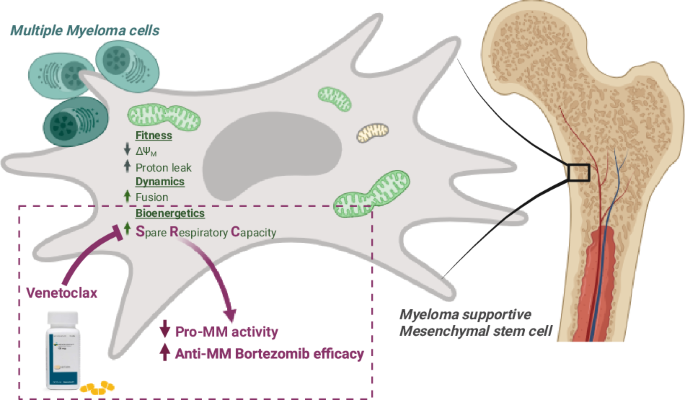
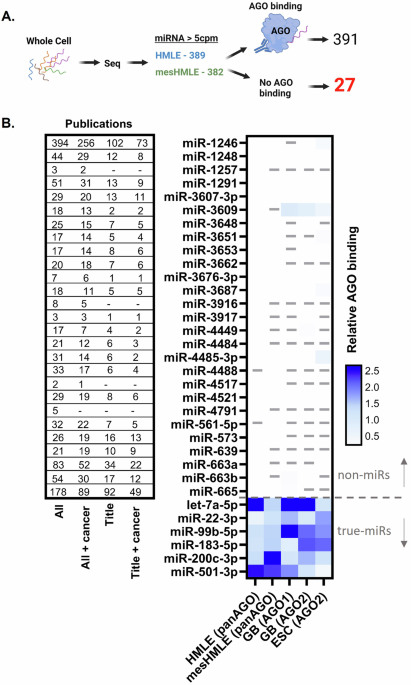
Comments (0)