Mice
Wild-type C57BL/6 N adult male mice and MR K/O adult male mice, 6 to 8 weeks old, were purchased from the Centre for Comparative Medicine Research (CCMR) of the University of Hong Kong. The animal facility is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. The mice were housed in a controlled environment with a 24-hour light/dark cycle and provided with access to food and water. The experimental protocols involving the mice were reviewed and approved by the Committee on the Use of Live Animals in Teaching and Research (CULATR) at the University of Hong Kong (approval number 22–065). All procedures performed on the animals were conducted under anaesthesia using ketamine/xylazine by the approved CULATR protocol.
LL-37-induced mouse model of rosacea
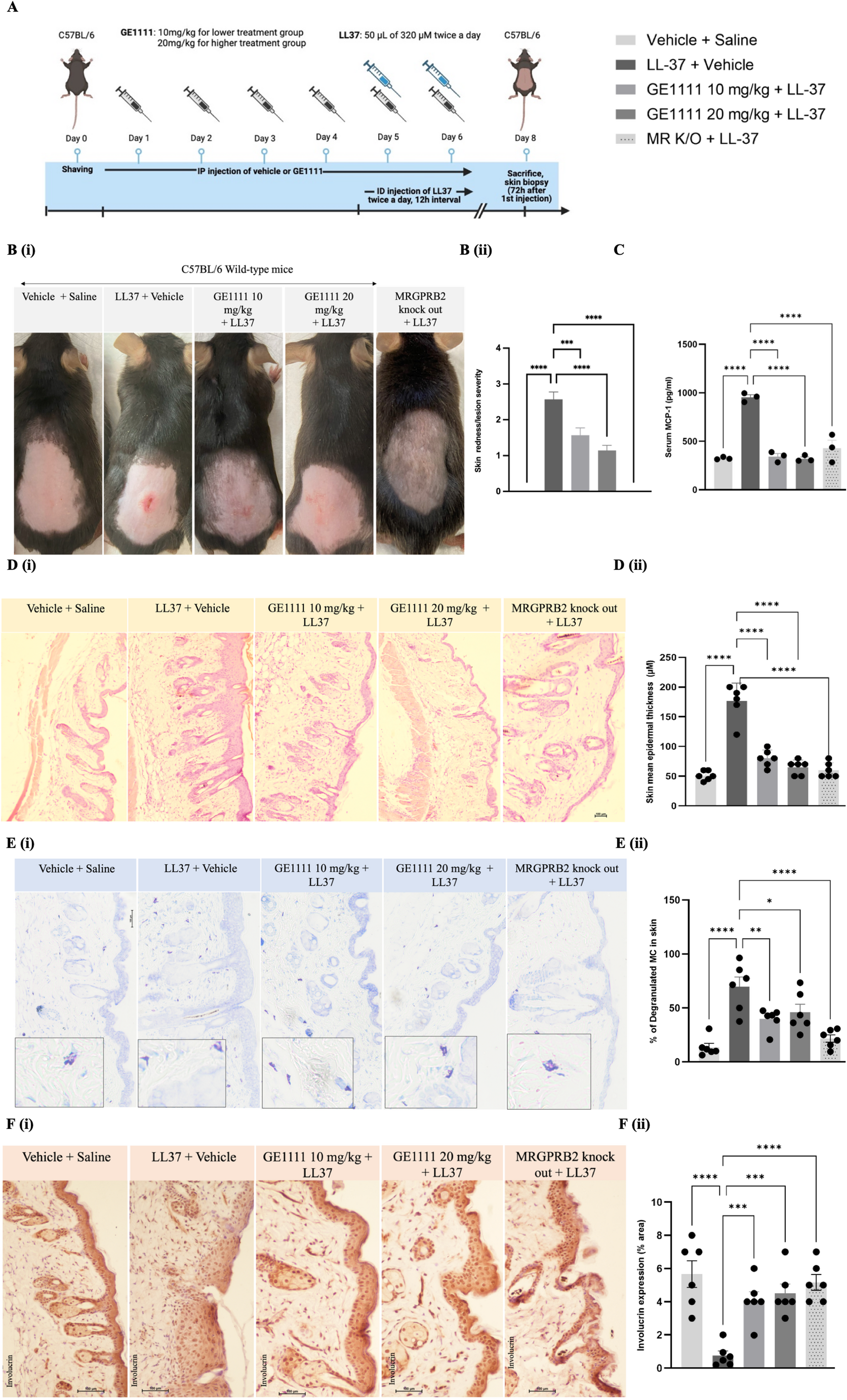
For the in vivo animal model, we used wild-type and MR K/O mice, divided randomly according to their body weight into 5 experimental groups (6 mice each). A schematic diagram outlining the experimental protocol is presented in Fig. 1A. The experimental groups were vehicle + saline (vehicle control), LL-37 treatment (disease control), lower GE1111 treatment group (T10), higher GE1111 treatment group (T20), and LL-37-MR K/O group. A total of 8 days of experimental protocol was followed briefly; on day 0, the dorsal skin area of experimental mice was shaved before LL-37/saline injection. From day 1 to day 6, 0.01% DMSO in PBS was injected via intraperitoneal injection to vehicle control, LL-37-induced disease control, and MR K/O groups. In contrast, GE1111 was injected intraperitoneally into T10 and T20 groups at a dose of 10 mg/kg and 20 mg/kg, respectively. On days 5 and 6, 50 µL of 320 µM LL-37 was injected intradermally twice a day with 12-hour intervals to LL-37 disease control, T10, T20, and MR K/O mice, while vehicle control mice were intradermally injected with vehicle. On day 8 (after 72 h of the first intradermal injection of LL-37), the severity of redness/skin lesions was graded as absent, mild, moderate, or severe (0–3). Mice were sacrificed, and the skin and blood were collected for downstream analysis (Fig. 2).
MRGPRX2/B2 agonist C48/80 induced mouse model of itch
We used wild-type C57BL/6 N and MR K/O mice for itch behavioral assay. Mice were acclimatised for 2–4 days before the experiment, and GE1111 (20 mg/kg body weight) was administered intraperitoneally 30 min before C48/80 injection. C48/80 (50 µL of 10 µg) was injected subcutaneously into the nape of the neck of mice, and scratching behaviour was observed for 1 h. A bout of scratching was defined as a hindpaw-directed continuous scratching movement at the C48/80 injection site. Scratching behaviour was quantified by counting the number of scratching bouts during the 1 h observation period and scored manually [20]. A schematic diagram of the experimental protocol is given on the left panel of Fig. 3B. C48/80 was selected as it activates MRGPRX2, mimicking non-histaminergic itch relevant to rosacea via mast cell degranulation [17, 20].
Enzyme-linked immunosorbent assay (ELISA)
The MCP-1 levels in the blood of the experimental mice were evaluated using the MCP-1 ELISA kit manufactured by Thermo Fisher Scientific (Catalog number BMS6005). The 4 times dilution was done for serum samples, and the assay followed the instructions.
Histology (H&E and toluidine blue staining)
Mouse skin and ear samples were fixed in 10% buffered formalin and were dehydrated using a series of ethanol concentrations ranging from 30 to 95%. Subsequently, these samples were embedded in paraffin blocks sectioned at a thickness of 5 μm and stained with H&E and toluidine blue staining according to our previous method [18]. Images of the sections were captured using a Nikon Eclipse Ni-U upright microscope equipped with a Nikon DS-Ri2 microscopic camera (Nikon, Tokyo, Japan) at both 10X and 20X magnifications.
Immunohistochemistry
Paraffin sections of the skin tissues were used to analyse the expression of the tight junction protein Involucrin. DAB Substrate Kit, Peroxidase (HRP), with Nickel (3.3′-diaminobenzidine), was used (SK-4100) for immunohistochemistry as per the manufacturer’s instructions. ImageJ software was used to quantitatively analyse the stained sections [21].
In vitro cell culture models
Chronic skin inflammatory conditions involve a complex interplay of skin and immune cells, such as MCs, keratinocytes, and macrophages. To mimic this environment in vitro, we designed and developed a model involving LAD-2 MCs, HaCaT keratinocytes, and RAW 264.7 macrophage cell line. LAD-2 cells were cultured in StemPro-34 medium supplemented with 10 mL/L Stem Pro nutritional supplements, penicillin-streptomycin (1:100), 2 mmol/L glutamine and 100ng/mL human stem cell factor and incubated at 37 °C in 5% CO2 incubator. The cell numbers were observed regularly, considering their doubling time. HEK-293, HaCaT and RAW 264.7 macrophage cell lines were kept in Dulbecco’s modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS) and penicillin-streptomycin (1:100). We looked at the crucial proteins and genes involved in rosacea via different techniques. To investigate the impact of MRGPRX2 on the interaction between MCs and other adjacent cells (keratinocytes and macrophages), we used a potent MRGPRX2 agonist LL-37 and novel small molecule MRGPRX2 antagonist GE1111. Briefly, HaCaT keratinocytes were seeded at a density of 3 × 105 cells/well (2 mL) in a six-well plate and 5 × 104 cells/mL (400 µL) in ibidi 8-well Chamber, removable slides (Catalog number: 80841). Cells were incubated overnight at 37 °C in 5% CO2 incubator. On the second day, LAD-2 cells were seeded at a density of 1.0 × 106 cells/well (800 µL) and treated with either 0.1% DMSO or 50 µM GE1111 (100 µL) for 30 min, followed by treatment with either naive media or 2.2 µM (10 µg/mL) of LL-37 (100 µL) for 2 h. After 2 h, the MCs were collected and centrifugated at 500 g for 5 minutes to isolate the MCs supernatant while MCs lysate was harvested for gene or protein expression analysis. The isolated supernatant (800 µL) was then transferred to keratinocytes and incubated for an additional 2 h. After 2 h incubation period, keratinocytes were harvested for subsequent gene or protein analysis. For immunofluorescence assay, 150 µL of LAD-2 MCs supernatant was transferred onto HaCaT keratinocytes and incubated for 14–16 h at 37 °C in 5% CO2 incubator.
Mast cell degranulation assay
We performed a β-hexosaminidase release assay according to our previously modified method [18]. For IC50 determination, MCs were treated with graded concentrations of GE1111 and stimulated with a fixed 90% effective concentration (IC90; ~5 µM) of LL-37. For EC50 determination, MCs were treated with a fixed concentration of GE1111 (5 µM) or buffer and stimulated with graded concentrations of LL-37 (0-100 µM). PNAG (4-Nitrophenyl N-acetyl-β-D-glucosaminide, 1.3 mg/ml) was added to each well of supernatant and lysate, and cells were incubated for 90 min at 37 °C (without CO2). The reaction was stopped by adding 50 µL of 0.4 M Glycine buffer. The extent of yellow color was measured at 405 nm using Victor 4X plate reader (PerkinElmer), and % MCs degranulation (% β-hexosaminidase release) was calculated.
Calcium flux assay
To evaluate and validate the effect of GE1111 on LL-37 mediated MRGPRX2 activation, we previously performed β-arrestin PRESTO TANGO assay. However, we were not able to find any change in the luminescence in the β-arrestin PRESTO TANGO assay for LL-37. Therefore, we designed a gene knock-in experiment by using a calcium flux assay in the MRGPRX2 transfected HEK-293 cell line. On day 1 of the experiment, HEK-293 cells were seeded at 1 × 106 cells per 100 mm cell culture dish overnight. On day 2, HEK-293 cells were transfected with MRGPRX2 DNA or mock-transfected using the Lipofectamine transfection reagent (Thermo Fischer Scientific) and incubated for 24 h. On day 3, MRGPRX2 transfected cells were transferred into 96-well black wall cell culture plates at a density of 2 × 104cells/well/80 µL (Thermo Fischer Scientific). On day 4, 40 µL of the 2X Fluo-4 NW calcium loading solution and or buffer was added (Molecular Probe, Invitrogen, USA). Cells were incubated for 30 min at 37 °C in a 5% CO2 incubator, followed by treatment with GE1111 (in the same manner as MC degranulation assay) and 30-minute incubation. After one hour of incubation, a graded concentration of LL-37 or buffer was added and the fluorescence intensity was measured at an excitation wavelength of 494 nm and an emission wavelength of 516 nm using Victor 4X plate reader (PerkinElmer) for 150 s. The ΔF was calculated by subtracting the maximum wavelength (after adding LL-37) from minimum fluorescence (before adding LL-37) and plotted by using Graph Pad Prism software 9.3.1.
Immunofluorescence assay
The experimental design was described in the above paragraph and in Fig. 4A. Briefly, HaCaT cells were incubated in the presence and absence of different MCs supernatant groups for a duration of 12–14 h. Fixed keratinocytes were blocked using 5% BSA for one hour at room temperature, followed by incubation with primary antibodies specifically targeting claudin-1 (Ab15098) and TSLP (PA5-89013) for 2 h at room temperature. Alexa Fluor 594 donkey anti-Rabbit IgG (red fluorescence) was used to detect claudin-1, and Alexa Fluor 488 donkey anti-Rabbit IgG (green fluorescence) was used to detect TSLP. The immunofluorescence staining was observed and captured using a Fluorescent Microscope equipped with a Nikon DS-Ri2 camera. Images were acquired at 20X magnificent levels to examine the cellular localization and expression levels of TSLP and claudin-1 within the HaCaT keratinocytes.
Western blotting
The in-vitro samples were subjected to lysis using RIPA lysis, extraction buffer, and cell scrapers, along with a phosphatase and protease inhibitor cocktail (Thermo-Fisher). The protein concentration of the samples was determined using the Bradford protein assay (Thermo-Fisher). To prepare the SDS-PAGE gels, 10%, and 15% acrylamide resolving gels (pH 8.8) were manually cast, with a 4% acrylamide stacking gel. Each well of the gel was loaded with 30 µg of protein, and the gels were run at 100 V for 2 h in a running buffer for gel electrophoresis. Primary antibodies specific to the proteins of interest, including STIM1 (CST-D88E10), TSLP (PA5-89013), claudin-1 (Ab15098), GAPDH (CST-141CO), phospho-ERK 1/2 (CST-4370T) and ERK 1/2 (CST-4695T) were prepared at a dilution of 1:1000 in 5% BSA and incubated with the membrane overnight at 4 °C. Following the primary antibody incubation, membranes were washed 3 times, 15 min each with TBS-T. Secondary antibodies such as Goat anti-rabbit IgG (CST-7074), diluted at 1:5000–10,000 in 5% BSA, were added to the membrane and incubated for 2 h at room temperature. After another 3 rounds of washing, immunoreactive proteins were detected using UVITEC Alliance LD with SuperSignal Technology. The band intensities were quantified and analysed using ImageJ software [21].
RNA isolation and quantitative RT‒PCR (RT-qPCR).
Alongside protein analysis, RT-qPCR was utilized to analyze the expression levels of various inflammatory cytokines, chemokines, and mediators in LAD-2 human MCs, HaCaT keratinocytes, and mouse skin samples. The RNA extraction process was carried out following the protocol of the FastPure Cell/Tissue Total RNA Isolation Kit V2 RC112 (Vazyme). In the case of mouse samples, additional homogenization was performed through agitation using the Fisherbrand™ Bead Homogenizer Accessory and Buffer RL from the FastPure Kit. Skin samples were first cut with sterilized scissors, then homogenized using an agitator for 20 min. To convert RNA into cDNA, the HiScript II 1st Strand cDNA Synthesis Kit R211 (Vazyme) was employed, following the instructions provided with the kit. Subsequently, PCR was conducted using the ChamQ SYBR Color RT-qPCR Master Mix (High ROX Premixed) Q441 (Vazyme) according to the kit’s protocol. The primers used for the PCR amplification, along with their corresponding sequences, can be found in Table 1.
Table 1 Primer sequence for human and mouse inflammatory cytokine genesStatistical analysis
Statistical analysis was performed using GraphPad Prism 9 software (GraphPad Software, La Jolla, California) or Microsoft Excel (Microsoft, Redmond, Washington) software. All the graphs were plotted by GraphPad Prism 9.3.1, and the data are presented as means ± SEMs of at least 3 independent experiments unless otherwise specified. The P values are represented by * and shown on top of the corresponding columns, as determined by 1-way ANOVA followed by Tukey’s multiple comparisons post-hoc test.



Comments (0)