
Remember me
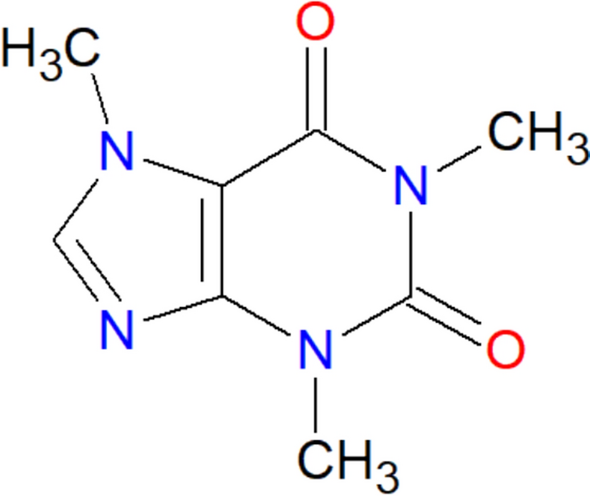


α-Glucosidase (Enzyme Commission [EC] no. 3.2.1.20), acetylcholinesterase (EC no. 3.1.17), tris–HCl, 2-naphthyl-α-d-glucopyranoside, 1-naphthyl acetate, and Fast Blue B were purchased from Sigma (St. Louis, MO, USA). Chloroform (pro analysis [p.a.]), water (mass spectrometry [MS] grade), ethanol (p.a. and liquid chromatography [LC] grade), methanol (LC and MS grade), formic acid (p.a. and MS grade), acetonitrile (LC and MS grade), and acetone (p.a.) were purchased from Merck (Darmstadt, Germany).
2.2 Biological materialWild seedy banana fruits were collected at the Central Highland region in Vietnam in December 2021: M. itinerans Cheesman in its ripe stage, M. acuminata Colla in its ripe stage, and E. glaucum Cheesman in its green stage (Fig. 1). The fruits were freeze-dried, and the seeds were then separated, ground to ≤ 0.5 mm with a Retsch-Ultra Centrifugal Mill ZM 200 (Haan, Germany), and kept at −80 °C for further analysis.
Fig. 1
Characteristics of the fruit of (a) Musa acuminata, (b) Musa itinerans, and (c) Ensete glaucum
2.3 ExtractionExtraction was performed on a Dionex ASE 350 (Thermo Scientific, Sunnyvale, CA, USA) with two different polar solvents in sequence: acetone (three times) and 70% aqueous methanol (three times). A sample of 4 g (± 0.01 g) of ground and freeze-dried seed powder (particle diameter < 0.5 mm) was placed in an extraction cell and extracted at 50 °C with a heating time of 5 min and a static time of 7 min. The extracts obtained from three cycles of acetone extraction were pooled in a flask (typically, the extract had a volume of about 160 mL). The solid residue was then extracted three times with 70% methanol under the same conditions, and the extracts were combined (160 mL). The solvents were evaporated on a rotary evaporator (Büchi, Flawil, Switzerland) in vacuo at 40 °C.
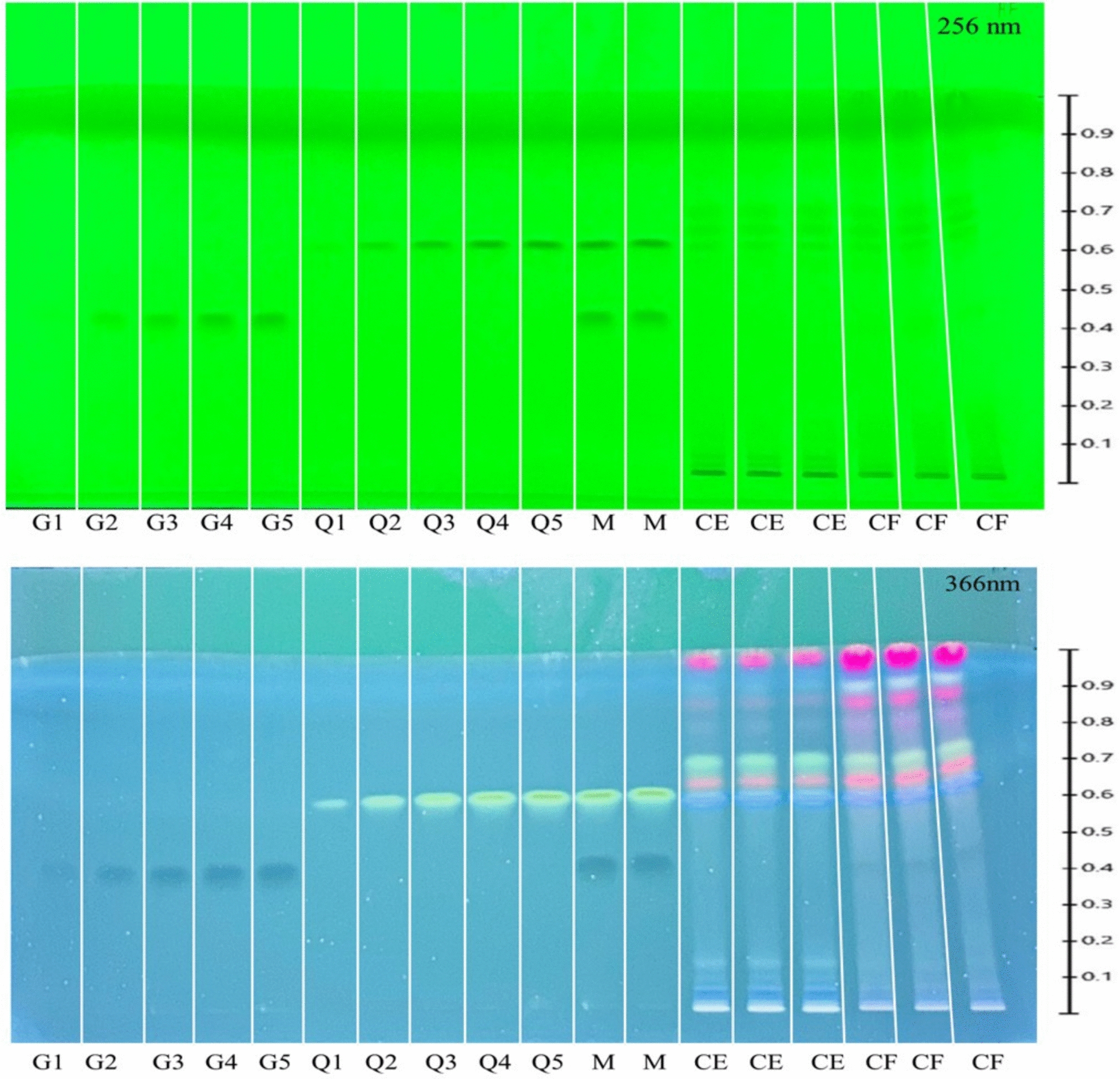
2.4 HPTLC‒direct bioautography2.4.1 HPTLC plate preparationHPTLC‒direct bioautography (HPTLC‒DB) was carried out on 10 cm × 10 cm HPTLC silica gel plates 60 F254 (Merck). Appropriate volumes of plant extract were sprayed as bands of 6 mm length onto the HPTLC plate using a Linomat 5 sample applicator (CAMAG, Muttenz, Switzerland). A mobile phase of chloroform‒methanol‒acetone‒formic acid (3:0.8:0.4:0.18, V/V) was used to separate the compounds. After separation, the solvent was evaporated on a hot plate at 70 °C for 10 min. The plate was then sprayed with Tris buffer (pH 10) for neutralization.
2.4.2 HPTLC‒DB acetylcholinesterase inhibitionThe assay was conducted following the protocol of Mroczek et al. [24] with some modifications. First, 1-naphthyl acetate (3 mL, 2 mg/mL in methanol) was sprayed onto the developed plate and dried. This was followed by spraying enzyme solution (3 mL, 5 U/mL in 50 mM tris–HCl buffer, pH 7.8, with bovine serum albumin, 1 mg/mL). The plate was incubated at 37 °C for 30 min in a humidity chamber (a tightly closed, compact chamber containing 50 mL of water) and sprayed with Fast Blue B (3 mL, 1.5 mg/mL in water). Acetylcholinesterase inhibitors are identified as colorless bands in a purple background.
2.4.3 HPTLC‒DB α-glucosidase inhibitionThe assay was performed according to the protocol reported by Galarce-Bustos et al. [25]. First, the developed plate was sprayed with substrate solution, 2-naphthyl-α-d-glucopyranoside (3 mL, 2 mg/mL in ethanol) and dried, followed by spraying with enzyme solution (3 mL, 4 U/mL tris–HCl buffer, pH 7.4 with bovine serum albumin, 1 mg/mL). The plate was incubated at 37 °C for 30 min in a humidity chamber. Finally, the plate was sprayed with Fast Blue B salt solution (0.5 mL, 1.5 mg/mL water). Glucosidase inhibitors are detected as yellowish zones on a purple background.
2.5 Active zone isolation and liquid chromatography‒tandem mass spectrometry (LC‒MS/MS)A 125-µL aliquot of the methanol extract of each species was applied as an 80 mm wide band to a 10 cm × 10 cm HPTLC plate with a Linomat 5 (CAMAG). After development, the targeted compounds that had shown inhibition in the preceding enzyme assays were located by their RF values and the fluorescence patterns at 366 nm. The targeted zones were scraped off, and the collected silica was extracted with methanol (200 µL) with ultrasonic treatment for 10 min at room temperature. After centrifugation at 2380 x g, the supernatant was filtered through a 0.22 µm filter and analyzed by LC‒MS/MS.
LC‒MS/MS analysis was carried out following the general conditions described by Carrasco-Sandoval et al. [26] for polyphenol analysis on an Arc Waters (Milford, MA, USA) ACQUITY UPLC I-Class PLUS device coupled to a photodiode array (PDA) detector and a Synapt XS mass spectrometer. Separation was carried out with a Phenomenex (Torrance, CA, USA) Kinetex XB-C18 (3.5 μm, 150 mm × 4.6 mm, 100 Å) column at 30 °C. The mobile phase was composed of water (A) and acetonitrile (B), both acidified with 0.1% (V/V) formic acid with the following gradient program: 0‒2 min: 20% B; 2‒6 min: 20‒30% B; 6‒14 min: 30‒70% B; 14‒15 min: 70% B; 15‒18 min: 70‒20% B; 18‒20 min: 20% B. The flow rate was set to 0.4 mL/min. MS analysis was performed with an electrospray ionization source in the negative mode using the following parameters: MS in continuum and resolution mode, m/z range 100‒1200, capillary voltage 0.8 kV, sampling cone 20 V, source temperature 120 °C, desolvation temperature 400 °C, cone gas 20 L/h, desolvation gas 600 L/h, and scan time 0.3 s. A collision energy ramp was set from 15‒45 eV for high energy and 4 eV for low energy with argon as collision gas. For m/z correction, a lock mass solution of leucine enkephalin ([M‒H]‒m/z: 556.2771) was applied in 30-s intervals during the sample acquisition. Mass calibration was performed using sodium formate (100‒1500 m/z). Data were acquired and analyzed by Waters Masslynx software (version 4.2.).
2.6 Purification and NMR analysisPurification of the targeted analytes (methanol extract of E. glaucum) was performed in two steps by flash chromatography (puriFlash XS 520Plus, Interchim, France) with ultraviolet/visible (UV/Vis) detection (200–400 nm). An initial normal-phase separation was performed with a silica gel column (Interchim SI-HP, 15 µm, 25 g) using the same solvent mixture as for the HPTLC separation, chloroform‒methanol‒acetone‒formic acid (3.0:0.8:0.4:0.18, V/V), at a flow rate of 6 mL/min. The fractions were assigned by HPTLC to the four targeted zones with RF values of 0.79, 0.65, 0.45, and 0.32. The fractions of interest were evaporated on a rotary evaporator in vacuo at 40 °C. Each target fraction was then fractionated again under reversed-phase conditions on a C18-column (Interchim PF-C18HP, 15 µm, 25 g). The used solvents were A (H2O with 0.1% formic acid) and B (acetonitrile with 0.1% formic acid). The flow rate was 8 mL/min. The elution was carried out with the following gradient that was suggested by the software controlling the flash chromatography system: 30% B (0 min) to 50% B (6‒10 min); 50% B (10 min) to 70% B (20 min) to 100% B (24 min). The separated fractions were evaporated in vacuo to obtain solids used directly for NMR analysis.
NMR spectra were recorded on a Bruker Avance II 400 (resonance frequencies 400.13 MHz for 1H and 100.63 MHz for 13C) equipped with a 5 mm inverse broadband probe head with z-gradients. Of the purified compound, 1.5‒7 mg was dissolved in 0.5 mL of methanol-d4 (99.8% D) and transferred into a 5 mm NMR tube. Standard 1H- and 13C-NMR spectra were recorded. Two-dimensional experiments, including homonuclear 1H/1H correlation spectroscopy (COSY), heteronuclear single-quantum coherence (HSQC), and heteronuclear multiple-bond correlation (HMBC) experiments using the standard Bruker pulse sequences, were performed for further analysis.
2.7 Compound identificationCassigarol E (2a), International Union of Pure and Applied Chemistry (IUPAC): 4-[(2R,3R)-2-(3,5-dihydroxyphenyl)-6-[(E)-2-(3,5-dihydroxyphenyl)ethenyl]-2,3-dihydro-1,4-benzodioxin-3-yl]benzene-1,2-diol or Maackin (2a′), IUPAC: 4-[(2S,3S)-3-(3,5-dihydroxyphenyl)-6-[(E)-2-(3,5-dihydroxyphenyl)ethenyl]-2,3-dihydro-1,4-benzodioxin-2-yl]benzene-1,2-diol. [M‒H]‒ = 485.1131. 1H-NMR (400 MHz, methanol-d4) δ (ppm): 7.14 (d, 1, J = 2.1 Hz, H-2), 7.07 (dd, 1, J = 8.4, 2.1 Hz, H-6), 6.97 (d, 1, J = 16.4 Hz, H-7), 6.95 (d, 1, J = 8.4 Hz, H-5), 6.86 (d, 1, J = 16.4 Hz, H-8), 6.66 (d, 1, J = 2.1 Hz, H-2′), 6.64 (d, 1, J = 8.2 Hz, H-5′), 6.47 (dd, 1, J = 8.2, 2.1 Hz, H-6′), 6.46 (d, 2, J = 2.2 Hz, H-10, H-14), 6.17 (t, 1, J = 2.2 Hz, H-12), 6.15 (t, 1, J = 2.2 Hz, H-12′), 6.10 (d, 2, J = 2.2 Hz, H-10′, H-14′), 4.74 (d, 1, J = 8.1 Hz, H-7′), 4.71 (d, 1, J = 8.1 Hz, H-8′). 13C-NMR (100 MHz, methanol-d4) δ (ppm): 159.7 (C-11, C-13), 159.3 (C-11′, C-13′), 146.7 (C-4′), 146.1 (C-3′), 145.5 (C-3), 145.0 (C-4), 141.0 (C-9), 140.1 (C-9′), 132.6 (C-1), 129.4 (C-1′), 129.0 (C-7), 128.4 (C-8), 121.0 (C-6), 120.7 (C-6′), 118.1 (C-5), 115.9 (C-5′), 115.8 (C-2′), 115.6 (C-2), 107.4 (C-10′, C-14′), 105.9 (C-10, C-14), 103.6 (C-12′), 102.9 (C-12), 82.3 (C-8′), 81.9 (C-7′). See Figs. S1‒S5 in the Supplementary Information for the spectra.
Scirpusin A (2b), IUPAC: 4-[3-(3,5-dihydroxyphenyl)-6-hydroxy-4-[(E)-2-(4-hydroxyphenyl)ethenyl]-2,3-dihydro-1-benzofuran-2-yl]benzene-1,2-diol. [M‒H]‒ = 469.1214. 1H NMR (400 MHz, methanol-d4), δ 7.03 (d, 2, J = 8.6 Hz, H-3, H-5), 6.83 (d, 1, J = 16.2 Hz, H-7), 6.75 (d, 1, J = 2.2 Hz, H-2′), 6.73 (d, 1, J = 8.2 Hz, H-5′), 6.64 (d, 2, J = 8.6 Hz, H-2, H-6), 6.63 (dd, 1, J = 8.2 Hz, 2.2 Hz H-6′), 6.62 (d, 1, J = 2.0 Hz, H-14), 6.55 (d, 1, J = 16.2 Hz, H-8), 6.25 (d, 1, J = 2.0 Hz, H-12), 6.17 (d, 1, J = 2 Hz, H-12′), 6.16 (d, 2, J = 2 Hz, H-10′, H-14′) 5.29 (d, 1, J = 6.2 Hz, H-7′), 4.33 (d, 1, J = 6.2 Hz, H-8′). See Figs. S6‒S8 in the Supplementary Information for the spectra.
Scirpusin B (3), IUPAC: 4-[(E)-2-[2-(3,4-dihydroxyphenyl)-3-(3,5-dihydroxyphenyl)-6-hydroxy-2,3-dihydro-1-benzofuran-4-yl]ethenyl]benzene-1,2-diol. [M‒H]‒ = 485.1217. 1H-NMR (400 MHz, methanol-d4), δ: 6.77 (d, 1, J = 16.4 Hz, H-7), 6.75 (d, 1, J = 2.2 Hz, H-2′), 6.74 (d, 1, J = 8.3 Hz, H-5′), 6.70 (d, 1, J = 2.0 Hz, H-2), 6.63 (d, 1, J = 8.3 Hz, H-5), 6.63 (dd, 1, J = 8.3, 2.2 Hz, H-6′), 6.62 (d, 1, J = 2.1 Hz, H-14), 6.57 (dd, 1, J = 8.3, 2.0 Hz, H-6), 6.54 (d, 1, J = 16.4 Hz, H-8), 6.25 (d, 1, J = 2.1 Hz, H-12), 6.17 (t, 1, J = 2.1 Hz, H-12′), 6.15 (d, 2, J = 2.1 Hz, H-10′,H-14′), 5.28 (d, 1, J = 5.9 Hz, H-7′), 4.33 (d, 1, J = 5.9 Hz, H-8′). 13C-NMR (100 MHz, methanol-d4) δ: 162.9 (C-11), 160.0 (C-11′, C-13′), 159.8 (C-13), 147.7 (C-9′), 146.6 (C-3/C-4/C-3′/C-4′), 146.5 (C-3/C-4/C-3′/C-4′), 146.4 (C-3/C-4/C-3′/C-4′), 146.3 (C-3/C-4/C-3′/C-4′), 137.0 (C-9), 135.0 (C-1′), 131.0 (C-1), 130.9 (C-7), 123.7 (C-8), 120.2 (C-6), 119.8 (C-10), 118.5 (C-6′), 116.4 (C-5), 116.3( C-5′), 114.1 (C-2), 113.7 (C-2′), 107.3 (C10′, C-14′), 104.4 (C-14), 102.2 (C-12′), 96.8 (C-12), 94.9 (C-7′), 58.1 (C-8′). See Figs. S8‒S13 in the Supplementary Information for the spectra.
Comments (0)