
Remember me





The ten notable studies demonstrated the use of spatial transcriptomics and/or proteomics to investigate H&N cancers, particularly HPV-positive SCC, de-novo SCC and SCC associated with OPMDs. However, they leveraged ST technology in the context of different study designs and objectives. Two studies [29, 30] applied ST to corroborate immune checkpoints identified a priori and studied via immunohistochemistry (IHC). This study design had the advantage that ST, which is expensive, could be applied to a smaller number of representative samples from the larger cohort. Seven studies applied ST to characterize gene expression across tissue types and/or the tumor microenvironment (TME) more broadly. Of these studies, one [31] investigated treatment resistance with a longitudinal case study, two [32, 33] investigated malignant transformation with a cross-sectional study design, which [33] also presents a spatial atlas, and four [34,35,36,37] investigated the role of spatial architecture in H&N SCC. The study by [35], in particular, trained an ML model on ST data to predict whether spots are at the “leading edge” of a tumor and then applied this model to different (non-H&N SCC) cancer types. The final notable study [38] was developed a cell-death-related risk score on publicly available bulk and scRNA-seq data and then generated ST for risk score validation. Overall, the ten notable studies identified gene expression signatures with prognostic and therapeutic implications, as discussed in more depth below.
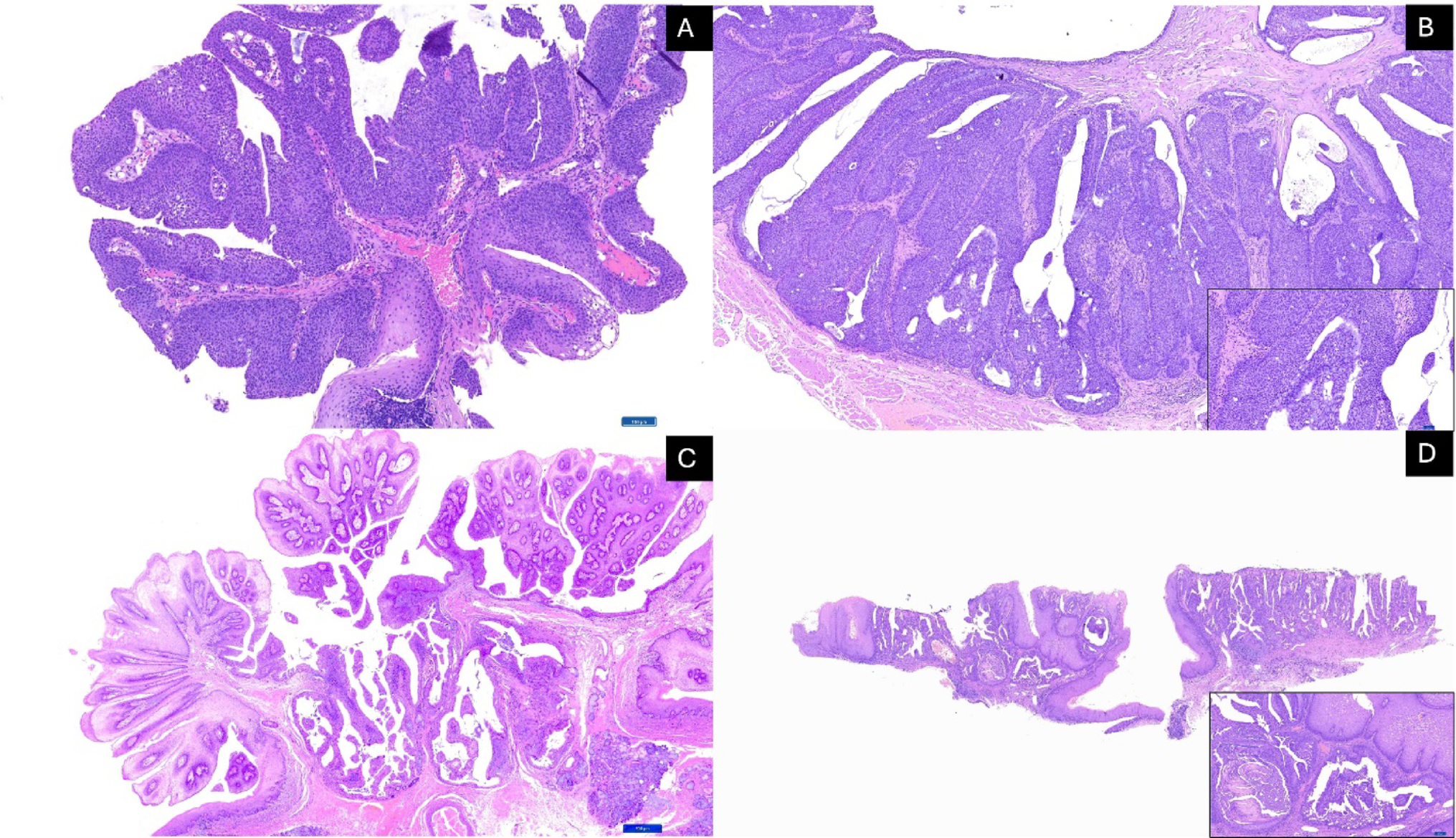
Noda et al. [29] utilized imaging approach to characterize the distribution and, in some regions, the mutual exclusivity, of immune checkpoint ligands (e.g., PD-L1 and B7-H4) within H&N SCC. The study involved IHC scoring of a large cohort. Transcript–protein concordance was validated by ST sequencing (Visium) of six representative H&N SCC paired samples, with squamous intraepithelial neoplasia and normal oral mucosa. Spatial mapping of CD274 expression corroborated immunostaining, showing the strongest enrichment in invasive H&N SCC regions, identified based on histology. ST also corroborated the presence of PD-L1 mRNA and protein expression in the PD-L1–high zones identified from the IHC, supporting the biological relevance of transcript-level measurements for checkpoint stratification.
Noda et al. [30] expanded upon their original study [29], investigating B7-H4 (VTCN1) as an alternative immune checkpoint target in H&N SCC, characterized by Low PD-L1 or immune-checkpoint-inhibitor-resistance. As in the group’s first paper, they combined ST (Visium) and multiplex IHC experiments on H&N SCC samples with paired squamous intraepithelial neoplasia and normal oral mucosa. Spatial maps revealed a mutually exclusive VTCN1–CD274 pattern in 83% of cases, with CD8A down-regulated in VTCN1-positive regions, indicating immune-suppressive niches. These transcriptomic findings corroborated IHC, showing reciprocal B7-H4/PD-L1 staining across tumor cores. Clinically, the authors proposed B7-H4 as a promising antibody–drug-conjugate target for PD-L1-negative or immune-checkpoint-inhibitor-resistant H&N SCC. Unlike the other studies that aimed to broadly characterize spatial patterns of disease progression, the two investigations by Noda et al. focused on evaluating an IHC marker that had been identified a priori. Because their goal was to validate this targeted IHC finding rather than explore global spatial gene-expression heterogeneity, their analytical pipeline appropriately relied on conventional differential-expression and pathway analyses rather than ML/DL based discovery approaches.
Iwasa et al. [31] used GeoMx digital spatial profiling to study spatial changes in gene expression and TME compartments from a single patient with SCC who acquired resistance to anti-PD-1 immunotherapy (nivolumab). Tumor samples collected pre- and post- resistance, along with normal tissue. The workflow involved selecting tumor, stromal, and normal regions of interest (PanCK ±), performing whole-transcriptome profiling, and comparing samples using differential expression and Reactome pathway enrichment within the GeoMx platform. These analyses revealed that, prior to immunotherapy, the initial tumor exhibited strong expression of antigen presentation and interferon-gamma signaling genes, whereas after resistance, these pathways were suppressed and replaced by dominant epigenetic reprogramming signatures including overexpression of PRMTs, HDACs, DNMT1, and EZH2 accompanied by downregulation of MHC class I molecules. Concurrently, the TME shifted from protein synthesis-related activity to G protein-coupled receptors and olfactory receptor signaling, indicating stromal reprogramming. Overall, these results suggest that immune escape in SCC might be driven by epigenetic suppression of antigen presentation and altered stromal signaling, highlighting potential therapeutic targets to restore immunogenicity and overcome resistance, although the conclusions of this study may be limited by the single-patient, observational design and possible therapy-specific effects.
Sun et al. [33] investigated the transformation of precancerous lesions to oral SCC. scRNA-seq and ST sequencing (Visium) was applied to biopsies from nine patients, including normal mucosa, dysplastic tissue adjacent to the malignant SCC lesions, and tumoral tissues. Segmentation of these regions was performed by a pathologist based on the histological images. Transcriptionally distinct zones were defined via unsupervised clustering and marker gene identification. Clusters were used in pathway enrichment analyses. Cell–cell communication was characterized with CellphoneDB [39]. These analyses identified initiation-associated epithelial and fibroblast signatures, immune and stromal cell reprogramming (especially immune‑inhibitory monocytes and pro-tumor signaling pathways such as VEGF), all spatially localized to precancerous zones. Validation, performed via functional modeling in mice, suggests that targeting the identified pathways (e.g., anti‑PD‑1, anti‑TGFβ) may prevent malignant progression, informing future early‑intervention strategies. Lastly, the study provided an integrative single-cell and ST atlas, capturing the cellular and molecular changes underlying SCC initiation from normal mucosa to dysplastic epithelium to early tumor. While this resource offers spatial resolution across disease stages which may or may not translate to longitudinal insights into the stepwise progression of SCC, as all samples were collected at a single timepoint. This contrasts with Iwasa et al. [31], who used a longitudinal study design.
Zhi et al. [32] also explored malignant transformation with a cross-sectional design, like the study by Sun et al. [33], but focusing on oral submucous fibrosis (OSF), an entity classified as a high-risk OPMD. ST sequencing (Visium) was applied to 4 OSF-SCC transformed samples and 1 conventional SCC sample (no OSF) as a control. Regions of interest, generated via unsupervised clustering, were annotated based on marker genes. After, differential gene expression and pathway analyses were conducted. The results were overlaid with spatial metabolomics data to assign metabolic programs to tissue zones, a unique aspect of this study. This analysis enabled the delineation of inflammatory stem-like cells (ISCs) undergoing partial epithelial–mesenchymal transition (pEMT) within the in-situ carcinoma region, eventually acquiring fibroblast-like phenotypes and participating in collagen deposition. This ISC > pEMT > CAF (Cancer Associated Fibroblasts) path mirrors biological transitions suspected in high-risk OPMDs. Importantly, the enrichment of polyamine metabolism and presence of tertiary lymphoid structures suggest both diagnostic and therapeutic relevance, supporting the utility of ST in identifying metabolic vulnerabilities and immunologic context. Due to the cross-sectional nature of this study, the proposed trajectory remains inferential, with causality suggested by spatial associations rather than demonstrated through longitudinal evidence.
Shaikh et al. [34] investigated the spatial distribution and role of fibroblast and myeloid cells subtypes driving lymph node metastasis. Their study analyzed H&N SCC samples from 23 gingivo-buccal SCC patients, applying both ST and IHC. After ST profiling (GeoMx) in selected regions of interest across tumor–stromal interfaces, spatial deconvolution (unsupervised learning) was to assess cell-type composition per spot. Standard methods for differential gene expression, gene set variation, and protein–protein interaction networks were applied. Overall, the analyses identified functional organization of tumor-stromal interfaces as a critical zone for metastasis progression. These are regions within 200 μm of the tumor edge, where intermediate fibroblasts, myeloid-derived cells, and neutrophils accumulate near tumor regions, while CAF and extracellular matrix genes (e.g., FN1 and COL5A1) localize toward stromal ends, especially in lymph node–positive cases. They observed that intermediate fibroblasts transition into CAFs and migrate outward, contributing to extracellular matrix remodeling and immune suppression, suggesting this spatial reorganization and stromal activation may promote an immune-suppressive microenvironment favoring cancer invasion and lymphatic spread. This study demonstrates the importance of spatial information in identifying potential checkpoints for inhibiting cancer metastasis, in concordance with the study by Zhi et al. [32].
Liu et al. [37] investigated the interplay between metabolic heterogeneity and the tumor immune microenvironment in oral SCC [24], although unlike Zhi et al. [32], who combined spatial metabolomics with transcriptomics to map polyamine metabolism and its association with inflammatory stem-like cell transitions in OSF-derived OSCC. Instead, Liu et al. leveraged scRNA-seq and ST (Visium), along with publicly available bulk and scRNA-seq data. Data integration enabled tumor immune microenvironment identification as well as per spot metabolic activity calculations with scMetabolism. Unsupervised clustering was performed on these spatial regions, and then clusters were classified into hyper-, normal-, or hypo-metabolic niches. Lastly, cell–cell communication was evaluated in these regions to delineate the coordinate work axis of epithelia cells, inflammatory CFAs, and regulatory T cells.
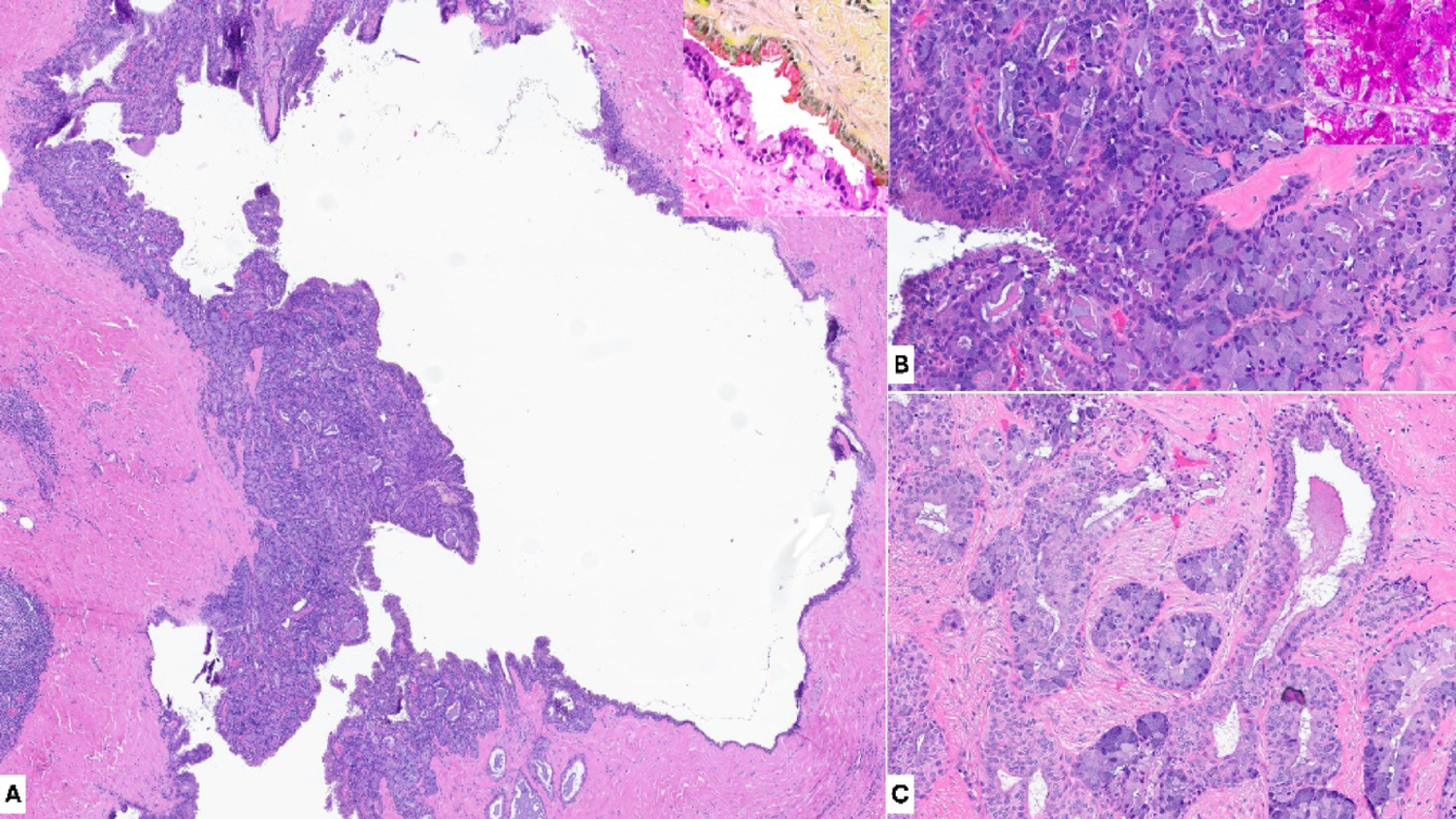
Arora et al. [35] investigated spatial architecture in oral SCC, identifying a transcriptionally conserved leading-edge zone associated with tumor aggressiveness and poor prognosis. ST sequencing (Visium) was applied to samples from 10 oral SCC patients. After unsupervised clustering, marker genes were used to label clusters as “tumor core”, “leading edge”, or “transitory”. Distinct and consistent spatial architectures between tumor core and leading edge were identified via differential gene expression, functional enrichment, pathway, and regulatory analyses [note that publicly available scRNA-seq data was leveraged for cell type deconvolution, as in Liu et al. [37]. RNA velocity and kinetic modeling was used to trace cell fate trajectories in the oral SCC HPV-negative samples, leading to the identification of gene signatures and differences associated with aggressive forms of oral SCC. Lastly, the annotated ST data was used to train models for predicting whether a spot was tumor core or leading edge. The application of these models to publicly available ST data for 17 different cancer types revealed that tumor core tended to be tissue-specific but leading edge had conserved gene expression profiles across cancer types, suggesting a potential common mechanism for tumor progression. This study marks an important step toward automating ST annotation through AI, demonstrating that learned architectural patterns can be transferred across tissue types and that spatial profiling can yield predictive spatial biomarkers. This zonal dissection could be directly applicable in OPMDs to stratify regions at highest risk of malignant transformation, although the limited number of samples used to train the model may limit the robustness of such inferences, underscoring the need for large, annotated ST data sets to enable reliable model training and generalization.
Zhang et al. [36] exclusively utilized publicly available data, making it distinct from the previously discussed notable studies. ST data were taken from the previously discussed study by Arora et al. [35] and scRNA-seq were data taken from [40] and [41]. Data integration was performed with the ML method RCTD [22]. This analysis revealed associations between epithelial cell subtypes and their interaction with normal-like CAFs and tumor invasiveness. Additionally, the identification of integrin and IGF1-mediated crosstalk provided mechanistic insight into epithelial-stromal interactions driving invasiveness, in concordance with Shaikh et al. [34]. This framework could be utilized to investigate other cell types and interactions associated with tumorigenesis in OPMDs, for example peri epithelial lymphocytes in OED have been reported to be significant for malignant transformation prediction [42]. Critically, this framework can utilize existing scRNA-seq or ST data, reducing the need for data generation, which can be prohibitively expensive for ST.
Pan et al. [38] also made use of publicly available data, like Zhang et al. [36]. Specifically, they leveraged bulk and ScRNA-seq data from both HPV-related and conventional oral SCC samples to train a consensus cell death-related risk score model. This risk score was validated with ST data (Visium) generated from tissue sections of laryngeal SCC from two patients and one normal larynx control patient. ST analyses revealed that high risk scores were also associated with TGF-β signaling pathways and that the interfaces between malignant cells and immune cells were enriched for high-risk scores. Unrelated to the ST validation, high risk scores were predictive of worse overall survival. Future studies could investigate the utility of such risks to OPMDs, which enable spatially resolved modeling of cell death, for early prediction of malignant transformation. Lastly, it is worth noting that this study evaluated whether learned patterns from one data generation modality translate another, similar to Arora et al.’s [35] who attempted transfer from oral SCC to other cancer types.
To summarize, the ten notable studies summarized above differ in their scope, analytical framework, and biological focus but are united by a common theme: spatial information is crucial for exposing the spatial interplay of cell types, signaling networks, and microenvironmental states that drive malignant transformation or cancer progression in OPMDs and H&N SCC. In many studies, joint ST and scRNA-seq data, often including data from public repositories, was also critical for combating trade-offs in spatial and molecular resolution [e.g. [35,36,37]]. Strong molecular signal (sequencing depth) in particular, is critical for robust inference of metabolic or immune activity. Another common theme is the growing emphasis on multi-modal data [e.g., ST and IHC [29, 30]] and multi-omics data [e.g., ST and spatial metabolomics; e.g. [32]]. Integrated data analyses provided complementary perspectives on tissue organization, revealing that the tumor–stromal interface is not a static boundary but a dynamic and interactive zone where fibroblast reprogramming, immune exclusion, and metabolic heterogeneity converge to drive malignant progression. These results underscore that understanding OPMD and H&N SCC progression requires viewing the tissue as a coordinated ecosystem rather than isolated cell populations.
Multi-omics and multi-modal studies naturally intersect with the growing use of ML and AI to further enhance the interpretive and predictive power of spatial data. The study by Arora et al. [35] illustrates how ML can extract latent spatial patterns and predict molecular phenotypes from high-dimensional ST datasets, whereas the study by Pan et al. [38] demonstrates models trained on non-spatial transcriptomic data can be used to stratify prognostic risk in ST. DL algorithms, in particular, hold promise for integrating histopathological imaging, gene expression, and clinical metadata to derive holistic, multi-modal representations of tumor biology, enabling scalable and automated insights into OPMD and H&N SCC progression.
Lastly, only one study leveraged a longitudinal design [31], and the only two studies to explicitly investigate malignant transformation from lesions [32, 33] used a cross-sectional approach. Longitudinal or temporally reconstructed spatial analyses may be needed to directly study how metabolic rewiring, stromal differentiation, and immune modulation unfold over time and to evaluate causality and directionality in disease progression. We expect this to be especially important in the context of OPMDs where malignant transformation is a slow and spatially heterogeneous process. This motivates us to present a conceptual framework for investigating malignant transformation of OPMDs.
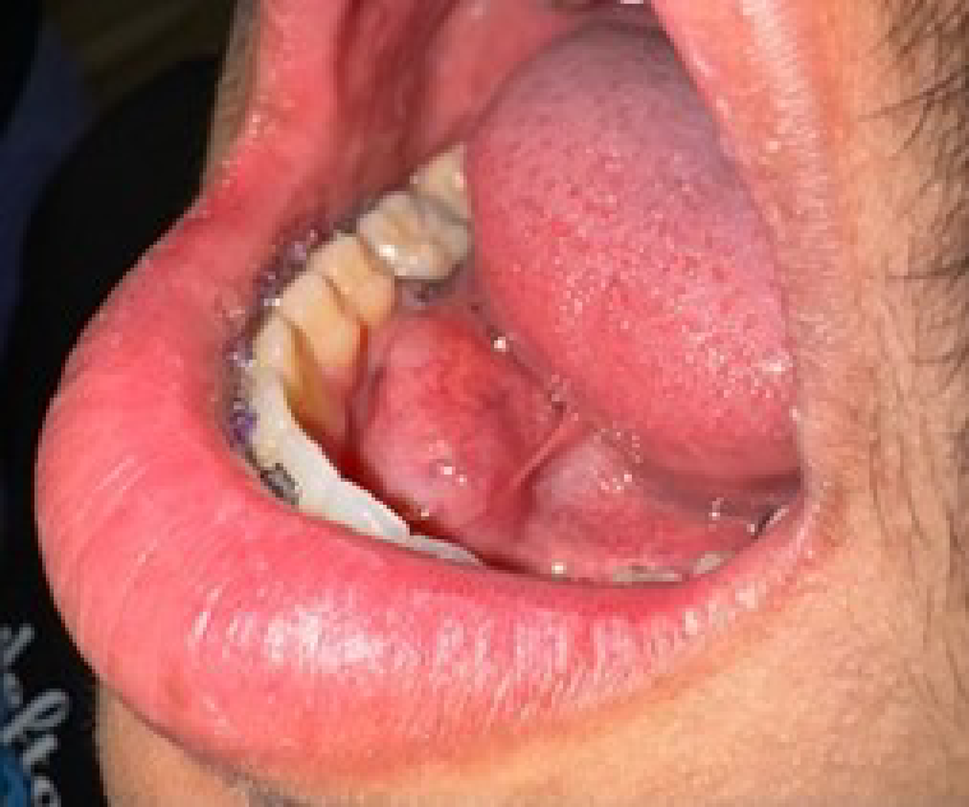
Conceptual framework for investigating and predicting risk in OPMDs malignant transformationStudy design and sample selectionA major challenge to study design is OPMD conditions are relatively rare, and biopsies are invasive and sometimes infeasible, creating challenges in curating sufficiently large and diverse datasets that are needed to power for downstream analyses. Longitudinal sampling is another critical yet challenging aspect of study design. Ideally such studies would involve temporal tracking by collecting serial biopsies taken from the same anatomical site in the same patient at multiple time points, to capture the evolving transcriptional landscape during transformation. However, prospective sampling techniques are often logistically and ethically restricted. A more practical and immediately feasible alternative is to leverage retrospective collection of archived Formalin-fixed paraffin-embedded specimens. Retrospective studies are greatly enabled by FFPE-compatible ST platforms, which enable spatial profiling of preserved tissue. With these technological advances, retrospective studies may enable temporal reconstruction of disease evolution and provide an attainable pathway for studying dynamic molecular changes leading to malignant transformation in OPMDs. However, retrospective studies will be fundamentally limited by the speed at which RNA degrades. Even with advanced preservation techniques, RNA can degrade quickly, leading to extremely poor ST quality data; thus, a critical first step in retrospective ST analyses is evaluation of RNA quality e.g. with RNAScope. Another critical consideration in applying ST to OPMDs is identifying retrospective cases that underwent malignant transformation within the same site that was initially biopsied, indicated by long-term follow-up of the OPMD with clinical- and histopathology-proven diagnosis. Furthermore, within each tissue section, it is essential to precisely select regions of interest that include both tumoral tissue, zones of malignant transformation, and adjacent normal tissue. This is particularly important for identifying differentially expressed genes among disease tissues. Researchers must also account for technical constraints of the ST platform, for example, Visium HD slides allow for only two 6.5 × 6.5 mm capture areas per slide which limits the amount of tissue that can be included.
Lineage tracingRegardless of the longitudinal study design, the future trajectories of biopsies lesions are unknown. Cell lineage tracing [43, 44], in conjunction with ST [45, 46], present a promising path forward to studying cellular trajectories across time and space. This technology, which is currently only leveraged within the context of xenographs [47] and mouse models [43, 44], could be used to spatial validate findings from traditional study designs. The idea is to apply genome editing, for example CRISPR Cas systems, to induce heritable mutations at target sites in the genome over time. The induced mutations can be captured via scRNA-seq or ST, along with gene expression data, and then used to reconstruct cell lineage trees. Leaves of the tree are labeled by cells or spots, the case of ST, have associated gene expression data, enabling spatial–temporal analyses of gene expression evolution [48, 49], cell state trajectories [50] and cell differentiation [51]. This approach, without ST, has been successfully used to study tumor evolution and metastasis in mouse models [43, 44]. Despite these exciting advances, lineage tracing technologies are still in their nascency. It is currently difficult to ensure the phylogenetic signal is sufficient for reconstructing an accurate and sufficiently resolved cell lineage tree and there can be data quality challenges as in non-lineage tracing ST studies [52].
Utilization of AI methods and risk stratificationAI models are commonly used for ST analyses, but another important direction of research is their application for stratifying malignant transformation risks, providing an objective transformation prediction tool that can be leveraged in the clinic. Several studies suggest that this is feasible. Two have employed DL-based image segmentation to analyze clinical images of various types of OPMDs [53, 54], demonstrating its applicability to classification different OPMD subtypes and prediction of malignant transformation rates, after incorporating clinical metadata [53, 54]. Relatedly, DL models trained and applied on scanned WSIs can detect subtle histopathological features associated with an increased risk of malignant transformation [42, 55]. Such models could serve as an aiding tool to alert pathologists of the potential of malignant transformation. We anticipate that combining ML/DL approaches with multi-model data, from clinical imaging, histopathology, and spatial omics, will further improve malignant transformation prediction, and ongoing advances in AI and multi-modal data integration indicate that an implementation is becoming increasingly feasible.
Workflow figureWe present an idealized conceptual workflow illustrating the potential integration of AI and ST technologies (Fig. 3) for future research designs investigating malignant transformation in OPMDs. This framework is intended to represent an idealized pipeline that highlights how these methodologies could be systematically combined to investigate spatially resolved molecular mechanisms and predict malignant transformation risk. Many challenges may be encountered while establishing these techniques which are further elaborated in the next discussion section. While a fully integrated approach has not yet been implemented, several of its individual components have already been successfully demonstrated in recently published studies applying AI models to histopathological image analysis and ST to characterize TME and predict genomic markers responsible for malignant transformation. Nevertheless, given the rapid evolution of these technologies, this model provides a realistic direction for near-future research once current limitations are systematically addressed. Emphasizing on the importance of incorporating both spatial and temporal resolution with retrospective longitudinal patient data, as well as in the integration of ST and AI approaches to enable alignment between distinct histomorphology layers and spatially defined gene expression clusters.
Fig. 3
An idealized workflow by utilizing explainable AI models on spatial multi-omics datasets from patient biopsies, with validation via mouse models and spatial lineage tracing, to provide novel insights into the pathogenesis and malignant transformation of OPMDs. Proposed workflow of OPMDs malignant transformation detection: A. Utilizing longitudinal patient’s data by retrieving oral potentially malignant disorders (OPMDs) samples of transformed and non-transformed cases for detecting malignant transformation via different modalities including: B. The use of digital pathology by developing a deep learning (DL) models that detects different forms of OPMDs clinically and histopathologically by digitizing histopathology slides generating high resolution whole slide images (WSI) that enable pathologists to virtually annotate different histopathological features related to malignant transformation and superimpose IHC images to develop a DL model with XAI features that detects malignant transformation rate. C. After WSIs used as input data to train a DL model to risk stratify high- versus low-risk lesions and identify features associated with progression and transformation. Here we highlight an example of the model’s output by detecting high risk features and digital immune scoring for risk stratification of different OPMDs particularly in those that have host immune lymphocytic response act as “neoantigen”. D. Patient’s FFPE tissue blocks are used for spatial transcriptomics experiments, which will lead to biomarker discovery of significant genes playing role in pathogenesis and transformation. The discovery can be validated via mouse models and spatial lineage tracing
Limited use of spatial transcriptomics and machine learning in current head and neck pathology research, challenges, and future directionsThe number of studies applying ST to OPMDs is quite limited to date. Our review identified just 10 studies that met the inclusion–exclusion criteria. This research predominantly focused on characterizing the TME, rather than the molecular events driving malignant transformation. A more detailed investigation into the spatial and transcriptional changes that occur as OPMDs progress to invasive cancer is essential for identifying early biomarkers and potential intervention targets.
Among the 10 included papers, only two trained ML models, although some incorporated ML-based tools for downstream analysis or evaluation, these were used solely for inference. To the best of our knowledge, none of the studies employed DL methods, of which there are many; we refer interested readers to Zahedi et al. [56] for a review of the many DL frameworks for ST analyses, including spatial clustering, gene imputation, and cell type deconvolution showcasing their utility in improving resolution and interpretability of ST data. Recent studies of other cancer types have demonstrated the potential of combining ST with DL to characterize the TME [57, 58], resolve cellular heterogeneity, and delineate key oncogenic pathways [59]. In particular, Ritter et al. [58] recently demonstrated the power of a graph-based DL framework for central nervous system tumors, achieving near-perfect tumor classification and virtual inference of IHC and copy number variation profiles. This type of integrated modeling demonstrates the transformative potential of DL approaches for H&N pathology diagnostics, enabling comprehensive molecular characterization from ST data while maintaining high accuracy. Applying such advanced methodologies to OPMDs could yield similar benefits, improving early detection and prediction of OPMD, particularly in settings where conventional histopathology may be insufficient.
The included studies also highlighted the importance of multi-modal data integration, combining ST with IHC, spatial metabolomics, and clinical metadata. Integrating diverse data types, including histological images, is critical for providing a comprehensive view of tumor progression unattainable from a single modality. Multi-model data also enables cross-modal inference, where information from one data type can be used to impute or enhance another. For instance, Rahaman et al. [60] developed a CNN-based model to impute transcriptomic profiles from H&E-stained WSIs in breast cancer, illustrating how computational approaches can augment ST capabilities. This framework is particularly valuable when molecular resolution is sparse (i.e., low coverage) to the trade-off in spatial-molecular resolution and/or degraded tissue quality. When such image-based predictions are combined with experimentally measured ST data, they enable cross-validation of findings where morphology-predicted molecular states derived from imaging can be verified against direct ST measurements, and ST-derived signatures can, in turn, guide interpretation of histological patterns. Such multi-modal integration is particularly advantageous for OPMDs, where understanding the relatio
Comments (0)