
Remember me
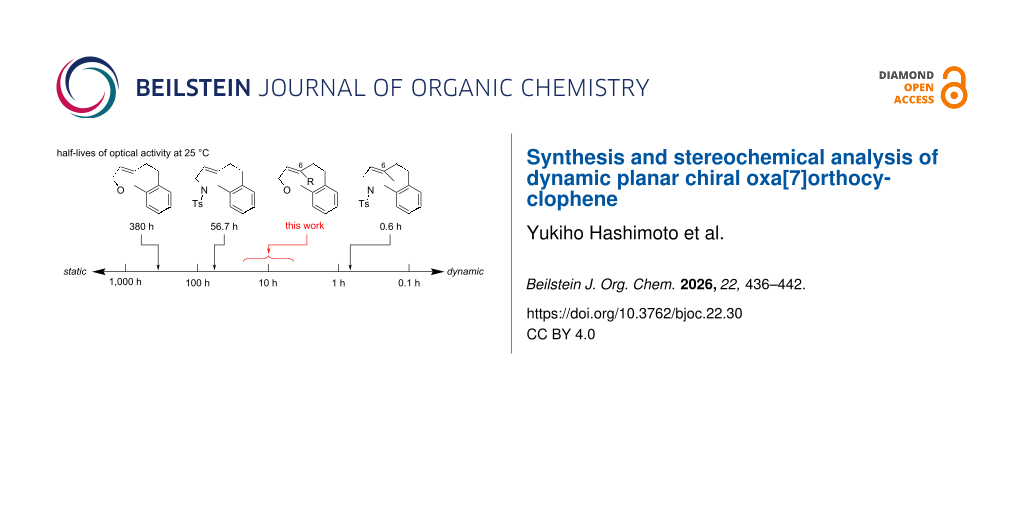




In the course of our study on planar chiral medium-sized cyclic molecules , we previously synthesized hetera[7]orthocyclophenes 1 having a heteroatom-embedded ansa-chain (X = O or NR) and an (E)-alkene moiety (Figure 1) , and found that the orthocyclophenes exhibit dynamic planar chirality in a wide range of stereochemical stability depending on the differences of the heteroatom and substituents on the (E)-alkene moiety . The half-lives of the optical activity optt1/2 of 1aa (X = O, Y = H) and 1ba (X = NTs, Y = H) were 380 h and 56.7 h at 25 °C, respectively, indicating that the stereochemical stability of the oxygen-containing cycle is higher than that of the nitrogen cycle: optt1/2 is one order of magnitude longer. On the other hand, C6-methyl-substituted nitrogen cycle 1bb (X = NTs, Y = Me) exhibits highly dynamic planar chirality (optt1/2 = 0.6 h at 25 °C), indicating that a C6-substituent decreases the stereochemical stability: optt1/2 is two orders of magnitude shorter. This planar chirality is too dynamic to handle the enantioenriched form without loss of enantiomeric purity in standard experimental operations. Based on these trends of the stereochemical stability of the hetera[7]orthocyclophenes, we expected that C6-substituted oxa[7]orthocyclophenes could exhibit reasonable dynamic planar chirality with optt1/2 approximately 10 h at 25 °C, which is dynamic but can be handled maintaining the enantiomeric purity in careful experimental operations .
![[1860-5397-22-30-1]](https://www.beilstein-journals.org/bjoc/content/figures/1860-5397-22-30-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Stereochemical stability of hetera[7]orthocyclophenes. aHalf-lives of optical activity at 25 °C in hexane. bPredicted half-lives of optical activity at 25 °C based on the measured value at 5 °C in hexane/EtOH 80:20.
In this study, we have designed, synthesized, and analyzed C6-substituted oxacyclophenes 1ab (X = O, Y = Me), 1ac (X = O, Y = Ph), and 1ad (X = O, Y = I), in which 1ad was used as the key intermediate for the synthesis of 1ab and 1ac via Kumada–Tamao coupling with Grignard reagents (Figure 2).
![[1860-5397-22-30-2]](https://www.beilstein-journals.org/bjoc/content/figures/1860-5397-22-30-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Synthetic plan of 1ab and 1ac from 1ad.
Results and Discussion Retrosynthesis and synthesis of the C6-iodo-substituted oxacyclophene 1adWe planned to construct the nine-membered cyclic ether skeleton of 1ad in the final step by an intramolecular Williamson etherification of haloalcohol 2 (Scheme 1). The iodine-substituted Z-alkene moiety of 2 was planned to be introduced through a trans-selective hydroiodination of propargyl alcohol 3 and the alkyne moiety in 3 can be introduced by an alkynylation of the formyl group of aldehyde 4. Thus, we started this synthesis from O-protected 2-bromobenzyl alcohol derivative 5 for the introduction of the propanal moiety of 4 by a Heck reaction with allyl alcohol.
![[1860-5397-22-30-i1]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-30-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthesis of oxacyclophene 1ad.
The synthetic route for the C6-iodo-substituted 1ad is shown in Scheme 2. The Heck reaction of O-TIPS-protected 5a with allyl alcohol in the presence of a palladium catalyst provided the aldehyde 4a in 78% yield . The reaction of 4a with CBr4 and PPh3, followed by treatment with n-BuLi, afforded the terminal alkyne 6. Subsequent preparation of the lithium acetylide using n-BuLi, followed by treatment with formaldehyde, afforded propargyl alcohol 3a in 70% yield over three steps. The reaction of 3a with Red-Al generated a vinyl aluminum species, which was then treated with iodine to provide the iodinated Z-alkene 7 in 81% yield. The hydroxy group of 7 was substituted with chlorine using oxalyl chloride in DMF, and the TIPS group was removed by treatment with hydrochloric acid to afford the chloroalcohol 2a (74% yield in two steps). After several attempts to cyclize compound 2a via Williamson etherification, the treatment with iPrMgBr for preparation of the magnesium alkoxide at highly dilute conditions (10 mM) and sequential cyclization successfully proceeded to provide the C6-iodo-substituted oxacyclophene 1ad in 79% yield.
![[1860-5397-22-30-i2]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-30-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of C6-iodo-substituted oxacyclophene 1ad.
Synthesis of C6-methyl and C6-phenyl-substituted oxacyclophenesThe Kumada–Tamao coupling of compound 1ad with MeMgBr and PhMgBr in the presence of a catalytic amount of NiCl2(PPh3)2 proceeded smoothly to provide the C6-methyl-substituted 1ab in 79% yield and the C6-phenyl-substituted 1ac in 99% yield (Scheme 3) . Suitable single crystals were obtained from racemic 1ac, and its X-ray crystallographic analysis was performed (CCDC 2513894). The solid-state structure of 1ac shows that the phenyl group on the E-alkene is directed antiparallel to the fused benzene ring. The dihedral angle of the alkene moiety (∠C4–C5–C6–C7) of 1ac is 146.4°, which is distorted by 33.6° from an ideal alkene plane.
![[1860-5397-22-30-i3]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-30-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of derivatives 1ab and 1ac, and ORTEP drawing of 1ac (ellipsoid set at 50% probability level).
Stereochemical analysis of C6-substituted oxacyclophenesNext, HPLC analyses using a chiral stationary phase (chiral column) of the C6-substituted oxacyclophenes were conducted. The baseline separations of the enantiomers of 1ab and 1ad were achieved by using CHIRALCEL OJ-H as the chiral column at 25 °C (Figure 3) .
![[1860-5397-22-30-3]](https://www.beilstein-journals.org/bjoc/content/figures/1860-5397-22-30-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Chromatograms of HPLC analysis of compounds 1ab and 1ad using an OJ-H column at 25 °C (OJ-H column, 4.6 mm × 250 mm, eluent: hexane/iPrOH 95:5, flow rate: 0.5 mL/min, monitoring wavelength: 220 nm).
In sharp contrast to 1ab and 1ad, the HPLC analysis of 1ac showed a plateau between the peaks of the enantiomers at 25 °C, indicating that the interconversion of the enantiomers proceeds on the separation time scale (Figure 4) . The interconversion plateau was still observed in the measurement at 5 °C, albeit only slightly.
![[1860-5397-22-30-4]](https://www.beilstein-journals.org/bjoc/content/figures/1860-5397-22-30-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Chromatograms of HPLC analysis of 1ac (IE column, 4.6 mm × 250 mm, eluent: hexane/iPrOH 95:5, flow rate: 1.0 mL/min, monitoring wavelength: 254 nm).
The phenyl-substituent effect on the stereochemical stability can be understood as a destabilization of the ground state due to the transannular repulsion, and a stabilization of the transition state for racemization by a conjugation effect of the phenyl group with the distorted alkene for a jump-rope rotation-type ring-flip (Figure 5) .
![[1860-5397-22-30-5]](https://www.beilstein-journals.org/bjoc/content/figures/1860-5397-22-30-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Plausible phenyl-substituent effect on the stereochemical stability of 1ac.
Enantioenriched samples of compounds 1ab and 1ad were obtained by HPLC separation using preparative scale chiral columns. We evaluated the stereochemical stability of 1ab and 1ad at 25 °C using the eluted samples (solvent: hexane/iPrOH 95:5). The plot of ln a (a = |R − S|/|R + S|) versus time furnished a straight line, affording the first-order rate constant k of racemization. The half-lives of the optical activity of 1ab and 1ad are 10.8 h (ΔG‡ at 25 °C: 24.4 kcal/mol) and 13.6 h (ΔG‡ at 25 °C: 24.5 kcal/mol), respectively, which are within the predicted range of stereochemical stability (Figure 6).
![[1860-5397-22-30-6]](https://www.beilstein-journals.org/bjoc/content/figures/1860-5397-22-30-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Kinetic measurements for the racemization of 1ab and 1ad.
For the determination of absolute stereochemistry of 1ab and 1ad, we performed HPLC analysis using circular dichroism (CD) detector . The HPLC analyses with OJ-H column of 1ab and 1ad showed similar sense of CD signals, in which the first eluates were detected as a positive signal, and the second eluates detected as a negative signal using the monitoring wavelength at 254 nm (Figure 7). Moreover, the direct CD spectra measurements in a flow cell showed characteristic Cotton effects. The CD spectrum of 1ab showed a weak positive Cotton effect around 270 nm and a strong Cotton effect around 230 nm, and that of 1ad showed a weak negative Cotton effect around 290 nm and a strong Cotton effect around 250 nm. Each TD-DFT calculations for (R)-1ab and (R)-1ad showed good agreement in shape for the CD spectra of the first eluates, thus we determined that the absolute stereochemistry of the first and second eluates corresponds to the (R)- and (S)-isomers, respectively.
![[1860-5397-22-30-7]](https://www.beilstein-journals.org/bjoc/content/figures/1860-5397-22-30-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: HPLC analysis of 1ab and 1ad using a CD detector at 5 °C (OJ-H column, 4.6 mm × 250 mm, eluent: hexane/iPrOH 95:5, flow rate: 0.5 mL/min, monitoring wavelength: 254 nm): a1), a2) chromatograms monitored at 254 nm; b1), b2) CD spectrum of the first eluates in a flow cell; c1), c2) calculated CD spectra using TD-DFT calculations at the B3LYP level using the 6-311G(2d,2p) basis set for C, H, and O atoms and the SDD basis set with the corresponding effective core potential for iodine.
Transformation of planar chirality of 1ab to central chiralityThe planar chirality of the methyl-substituted oxacyclophene can be transformed into central chirality without loss of enantiomeric purity. For example, (S)-1ab obtained by chiral HPLC separation using an OJ-H column was treated with m-CPBA, in which (S)-1ab was smoothly consumed at 0 °C within 30 minutus to afford epoxide 8 in 61% yield (Scheme 4). HPLC analysis of the obtained product 8 using chiral column showed an enantiomeric purity of >99% ee. In this reaction, m-CPBA can only approach from the outside of the ring skeleton of 1ab, so (5R,6R)-8 should be obtained from (S)-1ab.
![[1860-5397-22-30-i4]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-30-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Epoxidation of (S)-1ab.
ConclusionIn summary, we synthesized C6-substituted oxa[7]orthocyclophenes and revealed their dynamic planar chirality. By using the C6-iodo-substituted oxacyclophene as a common intermediate, C6-methyl- and C6-phenyl-substituted oxacyclophenes were synthesized efficiently. The enantiomers of the iodo- and methyl-substituted oxacyclophenes are isolable yet interconvertible at ambient temperature. The phenyl-substituted oxacyclophene showed more pronounced dynamic planar chirality than the iodo- and methyl-substituted derivatives. The planar chirality of the oxacyclophene was successfully transformed into central chirality by epoxidation without loss of enantiomeric purity.
© 2026 Hashimoto et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.
Comments (0)