
Remember me
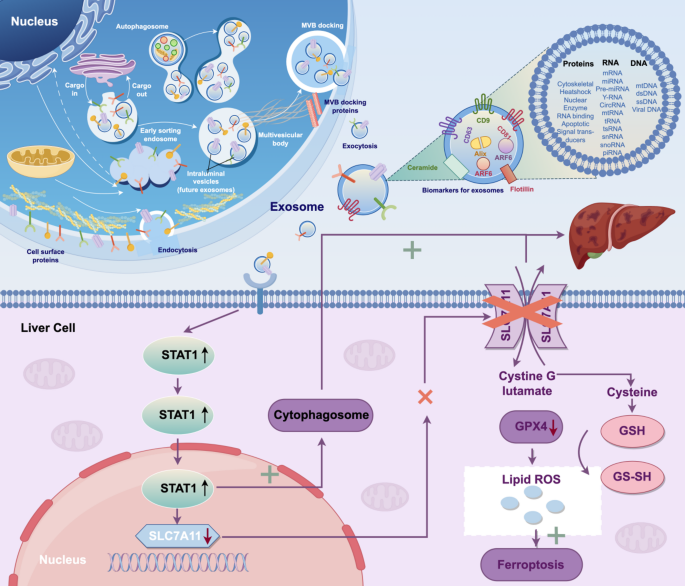

Peripheral exosomes have been implicated in the pathogenesis of multiple organ dysfunction during sepsis. However, their role in sepsis-associated liver injury (SALI) remains unclear. This study aimed to investigate the effects of circulating exosomes on hepatic injury and to elucidate the underlying molecular mechanisms of SALI. A murine sepsis model was established via intraperitoneal injection of lipopolysaccharide (LPS). Peripheral exosomes were isolated and co-cultured with murine hepatocytes (AML12 cells). RNA sequencing identified Signal transducer and activator of transcription 1 (STAT1) as a key regulator in exosome-induced liver injury. Since STAT1 functions upstream of the ferroptosis-related solute carrier family 7 member 11 (SLC7A11)–glutathione (GSH)–glutathione peroxidase 4 (GPX4) axis, further in vivo and in vitro experiments were conducted to clarify its mechanistic role. In vitro, exosomes derived from septic mice enhanced inflammatory responses in AML12 cells via STAT1-mediated autophagy and modulation of the SLC7A11–GSH–GPX4 axis, leading to ferroptosis. Inhibition of STAT1 abrogated these effects, whereas STAT1 overexpression potentiated them. In vivo, septic exosomes (sep-Exo) induced liver injury in mice, while suppression of STAT1 abolished the regulatory effects of sep-Exo on ferroptosis, autophagy, and hepatic inflammation. Our findings reveal a novel mechanism underlying SALI, whereby peripheral exosomes upregulate STAT1 to induce autophagy and modulate the SLC7A11–GSH–GPX4 axis, thereby promoting ferroptosis and hepatic inflammation during sepsis.
Graphical Abstract The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Peripheral circulating exosomes induce SALI by upregulating STAT1 to promote autophagy and modulating the SLC7A11-GSH-GPX4 axis to induce ferroptosis
Comments (0)