
Remember me
Cardiovascular diseases have been the leading cause of death worldwide, with coronary artery disease (CAD) being particularly prevalent and imposing significant health and economic burdens (GBD 2021 Causes of Death Collaborators, 2024). Atherosclerosis (AS) is the foundation for CAD and represents a persistent inflammatory condition. It is characterized by the buildup of various immune cells and lipids underneath the coronary endothelium that leads to luminal obstruction (Herrero-Fernandez et al., 2019). Once the endothelial function of the coronary wall is impaired, AS begins due to the accumulation of lipoprotein in the vessel intima. Endothelial cells (ECs) activate and trigger monocyte recruitment from the bloodstream. These monocytes differentiate into macrophages upon migrating into the subendothelial space and subsequently engulf lipids. The proliferation of foam cells results in fatty streaks that develop into atherosclerotic lesions. Vascular smooth muscle cells (VSMCs) are subsequently recruited, proliferate, and synthesize an extracellular matrix, promoting fibrous plaque formation. Ultimately, the encroachment of these plaques on the coronary artery lumen can lead to thrombogenic events (Badimon et al., 2012; Tabas et al., 2015). The progressive course of CAD is influenced by a combination of factors that complicates its management. Despite the existence of numerous clinical therapeutic strategies, the existing scientific knowledge does not fully explain the complex pathophysiological processes of CAD, and the treatment remains challenging (Bansal and Hiwale, 2023). Therefore, the opening up of novel therapeutic directions is a priority for current research.
In recent decades, epigenetics has become a new field of study that can potentially offer alternative strategies for treating human diseases (Portela and Esteller, 2010). Epigenetic changes, defined as heritable alterations in gene expression that do not affect DNA sequences, primarily involve DNA methylation, post-transcriptional histone modifications, and non-coding RNAs (ncRNAs) (Ragunathan et al., 2015). Epigenetic modifications remodel chromatin conformation to control DNA accessibility by modulating gene expression at multiple levels. Crucially, epigenetic processes are reversible and highly sensitive to environmental influences, making them attractive therapeutic targets (Baccarelli and Ghosh, 2012; Majnik and Lane, 2014; Marchio et al., 2019).
The involvement of epigenetics in the initiation and progression of CAD has become a major research focus. It has been reported that epigenetic changes are responsive to environmental risk factors associated with CAD and influence the progress of CAD by controlling the interaction between genes and the environment (Udali et al., 2013). For example, high-risk factors such as obesity cause CAD strongly associated with epigenetic pathways (Smail, 2019). CAD patients who smoke have specific DNA methylation patterns, while quitting smoking can reduce the risk of developing CAD (Steenaard et al., 2015). Besides, significant epigenetic alterations occur in the development of CAD, such as abnormal histone modifications in monocytes from CAD patients and a correlation between levels of circulating miRNA and the presence of CAD (Xiao et al., 2018; Kondracki et al., 2024). More importantly, epigenetic-based therapies may provide a new option for CAD patients. In fact, several existing pharmaceutical agents employed in the treatment of CAD have been shown to act through the modulation of epigenetic pathways. Other novel epigenetic drugs under development have initially shown promise for CAD patients (Bergonzini et al., 2023). Consequently, a comprehensive investigation of the epigenetic mechanisms implicated in the pathogenesis of CAD is crucial to advancing our understanding of its management.
2 Overview of epigeneticsDNA methylation, histone modifications, and non-coding RNA-based regulation are the main epigenetic characteristics of human cells (Egger et al., 2004; Rodenhiser and Mann, 2006; Wilson, 2008). Through epigenetic regulation, active gene states can be stably transmitted across cell generations, exerting a profound influence on cellular function and fate (Kelsey and Feil, 2013). This section provides an overview of these three regulatory mechanisms (Figure 1).
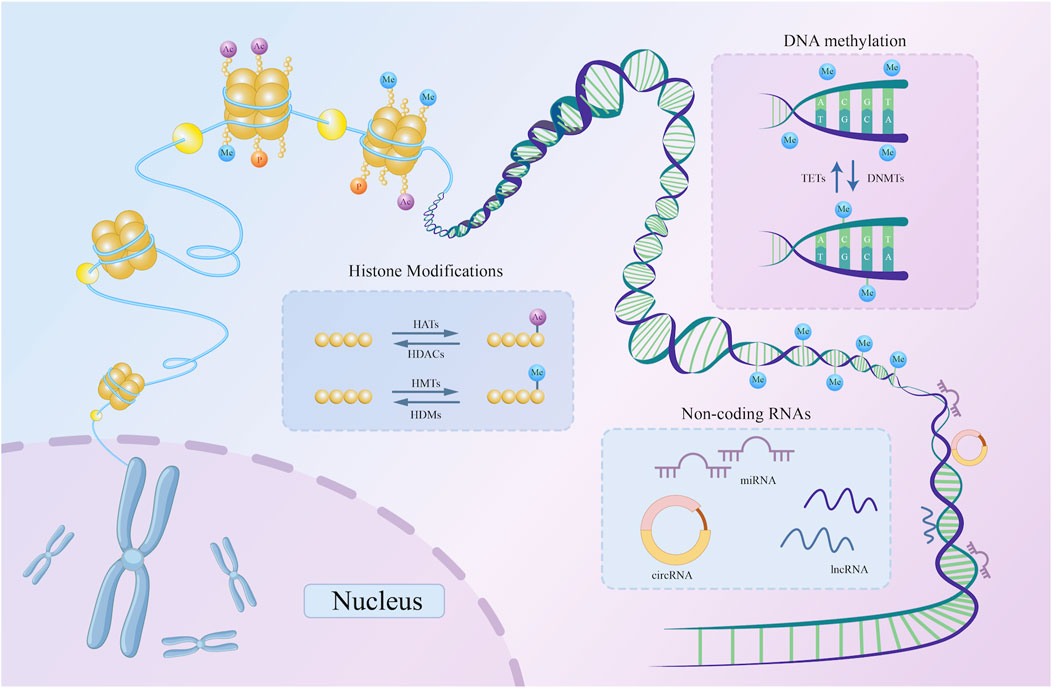
Figure 1. A schematic representation of three basic types of epigenetic regulation. DNMTs catalyze the addition of methyl groups to the 5′-CpG-3′ dinucleotide, while TETs catalyze the removal of methyl groups from the same dinucleotide. Histone acetylation and methylation are examples of histone modifications. HATs and HDACs catalyze the addition and removal of acetyl marks on the histone tail domain, respectively. Similarly, HMTs and HDMs are key enzymes catalyzing histone methylation. By attaching to DNA, non-coding RNAs, including circRNA, lncRNA, and miRNA, can alter the expression of particular genes. DNMT, DNA methyltransferase; TET, Ten-Eleven Translocation; HDAC, histone deacetylase; HAT, histone acetyltransferase; HMT, histone methyltransferase; HDM, histone demethylase; miRNA, microRNA; lncRNA, long non-coding RNA; circRNA, circular RNA; ncRNAs, non-coding RNAs; CpG, cytosine phosphate guanine.
2.1 DNA methylationDNA methylation, regarded as a major epigenetic alteration, has been extensively investigated as a pre-transcriptional modification. This process involves the covalent addition of methyl groups to the fifth carbon atom of cytosine within 5′-CpG-3' (cytosine phosphate guanine [CpG]) dinucleotide, forming 5-methylcytosine (Duan et al., 2016). DNA methyltransferases (DNMTs), such as DNMT1, DNMT3A, and DNMT3B, catalyze this reaction. DNMT3A and DNMT3B are responsible for de novo DNA methylation to create new methylation patterns, while DNMT1 maintains existing methylation markers (Chen and Zhang, 2020). Certain demethylating enzymes balance the changes in DNA methylation, such as Ten-Eleven Translocation (TET) enzymes, which remove methylation modifications from DNA by oxidizing methyl groups (Wu and Zhang, 2014).
In the human genome, approximately 70%–80% of CpG dinucleotides are predicted to be methylated. In contrast, demethylated CpGs are locally aggregated into CpG islands, rich in CpG dinucleotides with an uneven distribution. CpG islands typically reside in promoter regions and regulate transcription by modulating transcription factor binding, whereas methylated CpGs are primarily found in non-regulatory parts of the genome (Smith and Meissner, 2013; Greenberg and Bourc’his 2019). Methylation of promoter regions typically reduces accessibility and represses transcription, whereas hypomethylation is associated with active gene expression (Meehan and Stancheva, 2001; Curradi et al., 2002; Weber et al., 2007).
2.2 Histone modificationsHistones are alkaline proteins that package DNA into nucleosomes, forming the structural unit of chromatin. Closely linked to the function of DNA molecules as genetic material, histones are classified into five groups: H1/H5, H2A, H2B, H3, and H4. Except for H1/H5, the rest of the histones wrap DNA in an octamer. Each histone consists of an N-terminal tail domain and a C-terminal globular domain, where the tail domain is dynamically modified through acetylation, methylation, phosphorylation, and ubiquitination in response to cellular signals (Bhasin et al., 2006; Draizen et al., 2016). These modifications alter histones’ electrostatic charge and their binding capacity to DNA, contributing to conformational changes in the chromatin structure and influencing RNA transcription levels. Hence, post-translational histone modifications are significant mechanisms underlying gene control (Strahl and Allis, 2000; Berger, 2007; Rothbart and Strahl, 2014). Among these modifications, histone acetylation and methylation are particularly significant and are discussed in detail below.
2.2.1 Histone acetylationSince they contain several lysine and arginine residues, histones are positively charged. When acetylated on lysine residues, histones drift from DNA as their electric charge is neutralized, lessening the attraction between negatively charged DNA and the histone (Lee et al., 1993). The chromatin structure becomes loose, making it more susceptible to binding to transcriptional regulatory molecules. Therefore, acetylation promotes gene expression (Cheung et al., 2000). Furthermore, the balance of histone modifications is maintained by specific enzyme complexes that can dynamically add or remove these chemical groups (Yang and Seto, 2007). Along with transcription factors and co-regulators that serve as acetylation substrates, histone acetyltransferases (HATs) are recruited to acetylate lysine on histones during acetylation. Correspondingly, histone deacetylases (HDACs) can reverse this alteration by eliminating activating acetyl marks and restoring the positive charge (Blander and Guarente, 2004; Yang and Seto, 2008). The Gcn5-related N-acetyltransferases and p300/CBP families are prominent examples of HATs that are primarily localized in the nucleus. Furthermore, HDACs are classified into subgroups based on their structure and function, including class I HDACs (HDAC1, 2, 3, and 8), class II HDACs (HDAC4, 5, 6, 7, 9, and 10), class III HDACs (SIRT1–7), and class IV (Park and Kim, 2020). Collectively, HATs and HDACs add and remove acetyl groups from the N-terminal tails of lysine residues and are typically related to the activation and suppression of functional gene expression (Kaimori et al., 2016).
2.2.2 Histone methylationHistone methylation occurs predominantly at the lysine and arginine residues in histones H3 and H4, induced by histone methyl transferases (HMTs) (Wolf, 2009). The functional consequences of methylation are determined by the specific residues modified and the number of methyl groups added. The following methylation residues have been observed in histones H3 and H4: K4, K9, K27, K36, K79, and K20. Among that, H3K4, H3K36, and H3K79 allow transcription, while H3K9, H3K27, and H4K20 are repressive marks (Greer and Shi, 2012).
2.3 Non-coding RNAsAccording to bioinformatics statistics and the Encyclopedia of DNA Elements (ENCODE) project, approximately 70% of the human genome is transcribed into RNA, whereas only 1%–2% of DNA is directly involved in coding proteins. These protein-coding DNA sequences, known as exons, form the core of genes. In contrast, the remaining genomes that contain non-protein-coding regions, intronic regions, and repetitive sequences are translated into ncRNAs (Harrow et al., 2012). NcRNAs play essential regulatory roles within cells and participate in various biological processes, including gene silencing and activation, RNA splicing and editing, and protein translation (Kaikkonen et al., 2011; Cathcart et al., 2015). According to their size and mechanisms of action, ncRNAs are typically categorized into microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs) (Consortium, 2012; Quinn and Chang, 2016; Aufiero et al., 2019). Here, we provide a concise description of these three ncRNA types.
2.3.1 MicroRNAsMicroRNAs, approximately 20 nucleotides (nt) in length, have been identified as key regulators of gene expression across various biological networks. Their nucleotide sequences are highly conserved among species, highlighting their evolutionary significance (Friedman et al., 2009). To date, over 2,500 miRNAs have been identified, collectively regulating approximately two-thirds of protein-coding genes (Kozomara and Griffiths-Jones, 2014). miRNAs suppress gene expression by attaching themselves to the 3′untranslated regions of target mRNAs (Carthew and Sontheimer, 2009). Through complementary binding with mRNAs, miRNAs construct extensive regulatory networks that can reversibly adjust gene expression (Pillai, 2005). These miRNA-mRNA interactions are involved in most biological processes, endowing miRNAs with robust and effective regulatory capabilities to influence disease progression (Fabian et al., 2010).
2.3.2 Long non-coding RNAsLncRNAs were initially considered transcriptional ‘noise' without biological functions. However, recent research has revealed their extensive roles in gene regulation (Ma et al., 2015). Unlike miRNAs, lncRNAs are ncRNA molecules exceeding 200 nt originating from both the coding and non-coding regions of genes. They are expressed in the nucleus or cytoplasm and possibly released into extracellular fluids through extracellular vesicles (EVs) (Okazaki et al., 2002). LncRNAs exhibit high stability throughout an organism’s lifespan and demonstrate complex roles across different pathological states. Exerting their functions by forming RNA-protein interactions, lncRNAs can activate or inhibit pathways through interactions with transcription factors and modulating chromatin-modifying complexes (Quinn and Chang, 2016). In addition, lncRNAs interact with miRNAs in complex regulatory networks. For instance, incomplete base pairing allows miRNAs in RNA-induced silencing complexes to attach to target lncRNAs, which reduces the stability of lncRNA structure and function (Ballantyne et al., 2016). Furthermore, lncRNAs can influence target mRNA expression by competing with miRNAs directly, demonstrating their synergistic role to enhance and diversify biological information regulation (Ebert et al., 2007; Faghihi et al., 2010).
2.3.3 Circular RNAsCircRNAs, covalently closed-loop molecules, are typically between 100 nt and several kilobases. Predominantly located in the cytoplasm, circRNAs are present across different species (Memczak et al., 2013). CircRNAs often form closed-loop structures through back-splicing processes involving exon, intron, exon-intron, and tRNA intron regions of protein-coding genes (Zhang et al., 2013; Li et al., 2015; Chen et al., 2018). Due to the absence of both a 5′ cap and 3′ end tail, their circular configuration is more resistant to RNase degradation than linear ncRNAs. Certain circRNAs play protein-coding roles that differ from those of ncRNAs (Jeck and Sharpless, 2014). CircRNAs perform diverse functions in gene regulation. For instance, circRNAs interact with miRNAs as competing endogenous RNA (ceRNAs) and interfere with alternative splicing by intervening in looped exons and influencing the splicing patterns of mRNA precursors, resulting in changes in gene expression. CircRNAs can also bind to specific proteins to form complexes that modulate protein function by altering their conformation (Hansen et al., 2013).
3 Epigenetic modifications in CADEpigenetic alterations in the genome significantly impact the progression of CAD. Exploring the epigenetic mechanisms offers valuable insights into the pathogenesis of disease and aids in its diagnosis and therapeutic strategies. Specifically, epigenetic modifications occur in various cell types throughout the development of atherosclerotic lesions, including endothelial cells (ECs), vascular smooth muscle cells, and macrophages, which play a pivotal role in advancing AS (Grimaldi et al., 2015). Next, we discuss the role of epigenetic modifications in CAD separately in terms of the different cell types mentioned above.
3.1 Endothelial cell dysfunctionEndothelial cell dysfunction is recognized as the critical early event in the development of AS (Gimbrone and Garcia-Cardena, 2016). Under normal conditions, ECs generate vasodilator and vasoconstrictor molecules, such as endothelin and nitric oxide (NO), to regulate vascular tone and facilitate vascular repair, maintaining vascular homeostasis. However, different cardiovascular risk factors can trigger and exacerbate homeostasis disruption, resulting in loss of function (Sun et al., 2019; Xu et al., 2021). Figure 2 illustrates the specific epigenetic regulatory mechanisms underlying endothelial cell dysfunction.
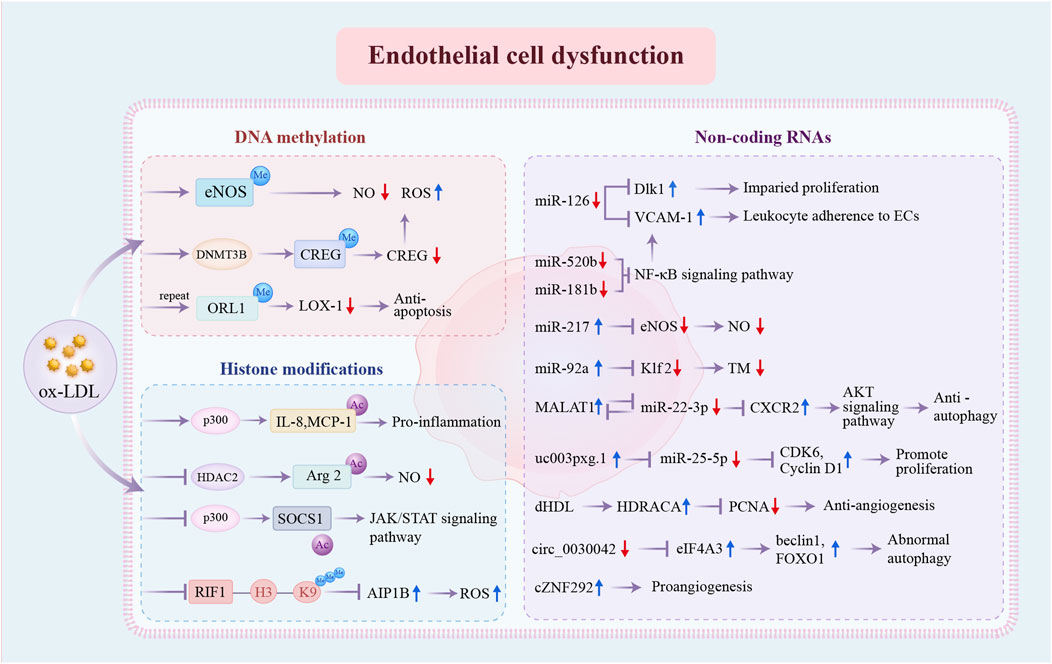
Figure 2. Epigenetic regulatory mechanisms involved in endothelial cell dysfunction. DNA methylation, histone modifications, and non-coding RNAs are the three distinct pathways of pro-atherosclerosis epigenetic regulation in ECs. DNA methylation: ox-LDL promotes DNA methylation at the eNOS promoter and increases DNMT3B expression and CREG promotor methylation. Repeated exposure to ox-LDL causes hypermethylation of OLR1 promoter regions, resulting in decreased LOX-1. Histone modifications: ox-LDL enhances the acetylated status of IL-8, MCP-1, and Arg2 and inhibits the acetylation of SOCS1 through the dysregulation of key enzymes p300 and HDAC2. Repressed H3K9 trimethylation of RIF1 promotes the expression of AIP1B. Non-coding RNAs: the expression levels of multiple non-coding RNAs have been changed, further influencing the expression of specific genes and ultimately resulting in EC dysfunction. ox-LDL, oxidized low-density lipoprotein; eNOS, endothelial nitric-oxide synthase; NO, nitric oxide; ROS, reactive oxygen species; CREG, cellular repressor of E1A-stimulated genes; ORL1, opioid receptor like-1; LOX-1, lipoprotein receptor-1; IL-8, interleukin-8; MCP-1, monocyte chemoattractant protein-1; Arg2, Arginase 2; SOCS1, suppressor of cytokine signaling-1; JAK/STAT, Janus kinase/signal transducer and activator of transcription; RIF1, Rap1 (RAS-related protein 1)-interacting factor 1; AIP1B, ASK1 (apoptosis signal-regulating kinase 1)-interacting protein-1B; Dlk1, delta-like 1 homolog; VCAM-1, vascular cell adhesion molecule 1; ECs, endothelial cells; NF-κB, nuclear factor kappa B; Klf, Krüppel-like factor; TM, thrombomodulin; MALAT1, metastasis associated lung adenocarcinoma transcript 1; CDK, cyclin-dependent kinase; dHDL, dysfunctional high-density lipoprotein; HDRACA, high-density lipoprotein-regulated angiogenesis in coronary artery disease; PCNA, proliferating cell nuclear antigen; eIF4A3, eukaryotic initiation factor 4A-III; FOXO1, Forkhead box O1; other abbreviations are shown in Figure 1.
3.1.1 DNA methylationDNA methylation is essential for the synthesis of endothelial nitric-oxide synthase (eNOS). Under physiological conditions, the eNOS promoter exhibits low levels of methylation in ECs, resulting in transcriptional permission. In contrast, ECs exhibit reduced eNOS expression levels, corresponding to DNA hypermethylation at the eNOS promoter in AS (Chan et al., 2004). In addition, the cellular repressor of E1A-stimulated genes (CREG), a vascular protective substance, is markedly downregulated in atherosclerotic vasculatures as oxidized low-density lipoprotein (ox-LDL) increases DNMT3B expression, leading to CREG promoter methylation. This is a novel mechanism that promotes EC dysfunction and the progression of AS (Liu et al., 2020). However, variations in the methylation status of pivotal genes in endothelial cells can also mediate adaptive protection against CAD. The lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1), encoded by the OLR1 gene, is the primary receptor for ox-LDL in ECs and facilitates pro-atherogenic effects (Sawamura et al., 1997). Unexpectedly, after repeated and continuous exposure to ox-LDL, ECs develop resistance to apoptosis due to epigenetic remodeling, which is associated with attenuated pro-apoptotic LOX-1 caused by modifications in the OLR1 promoter’s methylation (Mitra et al., 2011).
3.1.2 Histone modificationsThe accumulation of ox-LDL within the vascular vessel wall causes endothelial inflammation and vascular homeostasis imbalance, in which histone modifications affect gene transcription. HDAC2 is less expressed in human ECs because of ox-LDL, resulting in decreased endothelial nitric oxide via increased acetylation levels of Arginase 2 (Arg2) (Pandey et al., 2014). Ox-LDLs also induce specific modifications in interleukin (IL)-8 and monocyte chemoattractant protein-1 (MCP-1) gene promoters by attracting histone acetyltransferase p300 in ECs, causing ECs to release chemoattractants and proinflammatory cytokines (Dje N’Guessan et al., 2009). Meanwhile, regulation by proinflammatory cytokines suppresses RIF1 (Rap1 [RAS-related protein 1]-interacting factor 1)/H3K9 trimethylation-mediated epigenetic processes in ECs. This discovery leads to the identification of a shorter form of AIP1 (ASK1 [apoptosis signal-regulating kinase 1]-interacting protein-1) in diseased aortas of atherosclerotic plaques, with AIP1B augmenting reactive oxygen species (ROS) generation and further leading to EC dysfunction (Li et al., 2020). In addition, the suppressor of cytokine signaling-1 (SOCS1) is a negative mediator of inflammation and can alleviate endothelial injury. Nevertheless, its expression is reduced in CAD patients and in vitro because of the dysregulated acetyltransferase p300 (Xue and Deng, 2024). Collectively, these mechanisms lead to the activation of inflammation and EC dysfunction.
3.1.3 Non-coding RNAsThe expression of multiple miRNAs is usually repressed in the endothelium of CAD patients, thereby losing their protective effects. miR-126, which is highly expressed in ECs, regulates vascular inflammation by inhibiting the production of vascular cell adhesion molecule 1 (VCAM-1) (Harris et al., 2008). Disrupted laminar flow in AS reduces endothelial miR-126-5p levels, impairing endothelial recovery by upregulating delta-like 1 homolog (Dlk1), a Notch1 inhibitor (Schober et al., 2014). Additionally, the expression of miR-520b is suppressed in atherosclerotic plaques, which promotes EC inflammation and monocyte-EC communication via activation of the NF-κB p65-VCAM1 axis (Yang et al., 2021). miR-181b also acts as a critical inhibitor of NF-κB signaling, while its activity is impaired in AS (Sun et al., 2014). Conversely, several overexpressed miRNAs facilitate CAD progression. Aging-associated miR-217 is demonstrated to reduce NO production, promoting EC dysfunction and exacerbating AS in proatherogenic apoE−/−mice (de Yebenes et al., 2020). In addition, ECs subjected to disturbed flow exhibit increased miR-92a, which is dynamically regulated by shear stress. The transcription factor Krüppel-like factor 2 (Klf2), which is essential for preserving endothelial function, is less expressed when miR-92a is overexpressed (Wu et al., 2011).
LncRNAs also regulate endothelial integrity. The lncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) is upregulated in patients with unstable angina and protects ECs from ox-LDL-induced endothelial injury, partly by upregulating CXCR2 expression through competitive binding with miR-22-3p (Tang et al., 2015). Likewise, another study verified that EC proliferation was decreased by the genetic ablation of MALAT1 (Michalik et al., 2014). These two studies indicate that the upregulated lncRNA MALAT1 plays a protective role towards the endothelium in CAD. In contrast, patients with CAD have higher levels of the lncRNA uc003pxg.1, which binds to miR-25-5p to promote CAD development by increasing EC migration and proliferation (Li et al., 2021). Normal HDL suppresses the expression of lncRNA HDRACA in ECs to promote angiogenesis. However, when HDL from CAD patients undergoes several changes that cause it to become dysfunctional (dHDL), it loses its capacity to promote angiogenesis due to the presence of the lncRNA HDRACA (Mo et al., 2023).
Circular RNAs are emerging functional molecules in CAD development. Aberrant autophagic mortality of ECs is harmful to atherosclerotic plaques because EC loss encourages lesional clots. circ_0030042 is markedly downregulated in CAD, which promotes ox-LDL-induced abnormal autophagy in ECs and destabilizes atherosclerotic plaques by targeting eukaryotic initiation factor 4A-III (eIF4A3) (Yu et al., 2021). In the vascular endothelium, the hypoxia-induced circRNA cZNF292 is highly expressed and exhibits proangiogenic activity in vitro (Boeckel et al., 2015).
3.2 Macrophages and imbalanced lipid homeostasisAS is a persistent inflammatory condition of the artery wall driven by hyperlipidemia. The etiology of AS is heavily influenced by lipid metabolism, particularly cholesterol metabolism and homeostasis. During this process, cholesterol builds up in macrophage-derived foam cells, resulting in lipid accumulation within plaques, a hallmark of early-stage atherosclerotic lesions. Further clarification of the mechanisms leading to abnormal lipid metabolism and foam cell accumulation is vital for preventing plaque formation and rupture (Ouimet and Marcel, 2012).
An imbalance between low-density lipoprotein cholesterol (LDL-C) and high-density lipoprotein cholesterol (HDL-C) significantly contributes to the development of AS (Emerging Risk Factors et al., 2009). Epigenetic regulatory patterns have been identified in genes involved in lipid transport in CAD. ATP-binding cassette A1 (ABCA1) accelerates cholesterol transport from extrahepatic tissues to new HDL particles. The DNA methylation levels of the ABCA1 promoter are elevated in familial hypercholesterolemia, correlating with reduced circulating HDL-C levels and a higher risk of CAD (Guay et al., 2012). Additionally, several ncRNAs affect foam cell production by regulating macrophage cholesterol export via ABCA1. miR-33 and lncRNA growth arrest-specific 5 (GAS5) are highly expressed in CAD macrophages and suppress ABCA1 expression (Rayner et al., 2010; Meng et al., 2020). Furthermore, a novel lncRNA, CHROME, enhances cholesterol efflux and HDL synthesis by inhibiting the effects of a group of functionally similar miRNAs, such as miR-33 (Hennessy et al., 2019). miR-33 also drives lipid droplet accumulation in macrophages via a lysosome-dependent mechanism that represses key autophagy regulators (Ouimet et al., 2017). The scavenger receptor type B1 (SR-BI), predominantly expressed in the liver, serves as an HDL receptor. miR-223 inhibits HDL-C uptake in atheroprone mice by directly targeting and regulating SR-BI expression (Vickers et al., 2014). Similarly, miR-125b impairs macrophage-specific reverse cholesterol transport by targeting SR-BI (Hueso et al., 2022). In addition, miR-148a modulates hepatic low-density lipoprotein receptor (LDLR), a receptor responsible for clearing circulating LDL-C, thereby influencing cholesterol homeostasis (Goedeke et al., 2015). Together, these findings suggest that epigenetic regulation of lipid metabolism genes significantly influences circulating LDL-C and HDL-C levels, thus influencing the risk of AS.
Beyond regulating lipid metabolism, epigenetics regulates macrophage phenotype (Figure 3). HDAC9, abundantly expressed in atherosclerotic macrophages, promotes the inflammatory M1 macrophage phenotype, contributing to AS. HDAC9 deletion reduces inflammation via increasing acetylation of H3 and H3K9 at peroxisome proliferator-activated receptor (PPAR-γ) promoters in macrophages, decreasing AS in vivo (Cao et al., 2014a). Similarly, increased HDAC3 expression in atherosclerotic lesions is associated with inflammatory macrophages (Hoeksema et al., 2014). In contrast, ox-LDL-loaded macrophages negatively regulate proinflammatory gene expression at late stages, also regulated by epigenetic mechanisms such as HDAC activity. Macrophages develop the Mox phenotype, a special polarization state that is distinguished by increased production of antioxidant genes under the guidance of nuclear factor erythroid 2-related factor 2 (NRF2) (Jongstra-Bilen et al., 2017). Notably, this reduction in inflammatory response may act as a form of adaptation for foam cells, contributing to a persistent atherogenic state in the lesions. Thus, macrophage polarization is influenced by epigenetic changes, with distinct regulatory patterns emerging at different stages in AS.
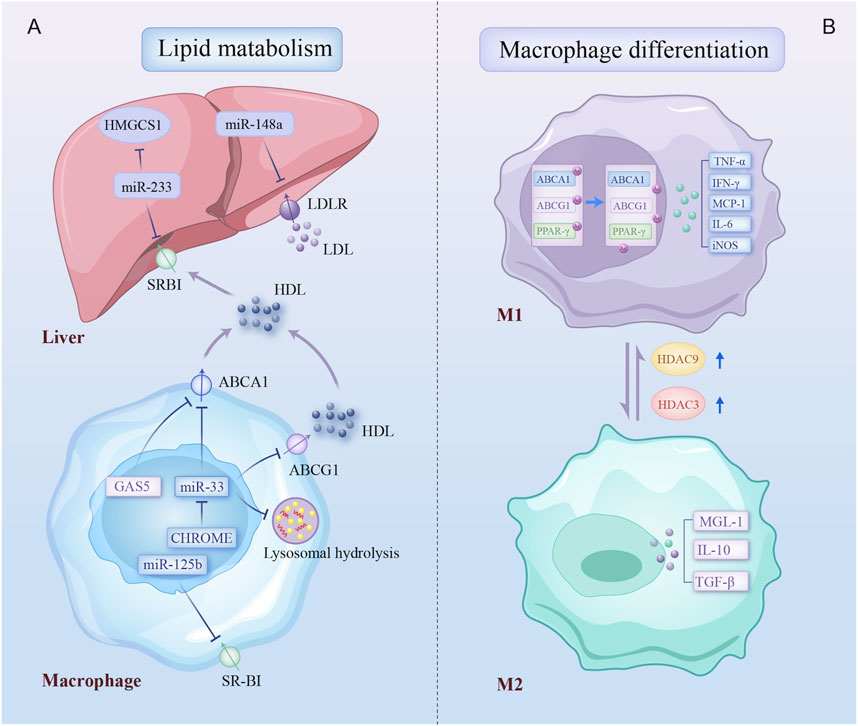
Figure 3. Epigenetic regulatory mechanisms involved in lipid metabolism and macrophage differentiation. Epigenetic modifications regulate two biological processes of macrophage: (A) multiple non-coding RNAs contribute to dysregulation of cholesterol production, uptake, and efflux by inhibiting key cholesterol transporters, the cholesterol-producing enzyme, and lysosomal-dependent autophagy processes in liver and plaque macrophage; (B) macrophage in atherosclerotic plaques differentiates into a proinflammatory phenotype due to increased expression of HDAC3 and HDAC9. Arrows on specific transporters indicate the direction of transport. HMGCS1, 3-hydroxy-3-methylglutaryl-CoA synthase 1; SR-BI, scavenger receptor type B1; LDLR, low-density lipoprotein receptors; ABCA1, ATP binding cassette subfamily A member 1; GAS5, growth arrest-specific 5; ABCG1, ATP binding cassette subfamily G member 1; CHROME, cholesterol homeostasis regulator of miRNA expression; PPAR-γ, peroxisome proliferator-activated receptor gamma; IFN-γ, Interferon-gamma; TNF-α, tumor necrosis factor-alpha; iNOS, inducible nitric oxidase synthase; TGF-β, transforming growth factor-beta; MGL-1, macrophage galactose-type lectin-1; other abbreviations are shown in Figures 1, 2.
3.3 Phenotype switch of vascular smooth muscle cellsIn the atherosclerotic lesion microenvironment, VSMCs dedifferentiate and proliferate. This aberrant phenotypic switching of VSMCs is a crucial step in the development of AS (Figure 4).
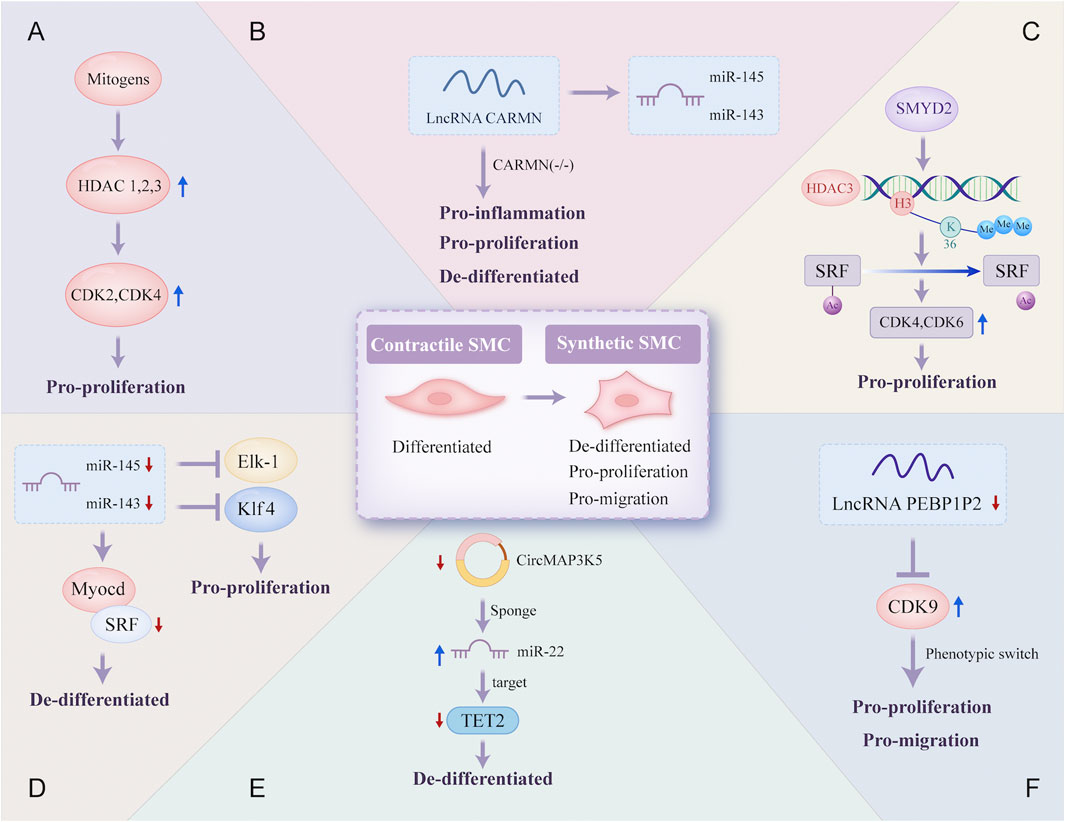
Figure 4. Epigenetic regulatory mechanisms involved in the phenotype switch of vascular smooth muscle cells. Contractile SMC switches to a synthetic phenotype that is de-differentiated, pro-proliferation, and pro-migration in atherosclerosis through a variety of different epigenetic regulatory mechanisms: (A) mitogens induce HDAC 1, 2, and 3 transcription in SMC, which promotes SMC proliferation due to increased transcriptional regulation of CDK2 and CDK4; (B) lncRNA CARMN can promote the expression of miR-145 and miR-143, and early loss of CARMN controls the phenotype switch of SMC; (C) the overexpressed SMYD2 promotes the expression of HDAC3 through H3K36 tri-methylation at its promoter, and then HDAC3 deacetylates SRF, finally promoting SMC phenotypic switching; (D) expressions of miR-145 and miR-143 are reduced, which allows the proliferation of VSMC through targeting transcription of SRF, Elk-1 and Klf4; (E) CircMAP3K5, significantly downregulated, promotes the de-differentiation of SMC by acting as a TET2-mediated competing endogenous miR-22-3p sponge; (F) the decreased expression of lncRNA PEBP1P2 distinctly promotes proliferation, migration, and phenotypic switching in SMC by directly binding to CDK 9. VSMC, vascular smooth muscle cell; Elk-1, ETS-like transcription factor; Myocd, myocardin; SRF, serum response factor; CARMN, cardiac mesoderm enhancer-associated non-coding RNA; CircMAP3K5, circular mitogen-activated protein kinase 5; SMYD2, SET (Suppressor of variegation, Enhancer of Zeste, Trithorax) and MYND (Myeloid-Nervy-DEAF1) domain-containing protein 2; PEBP1P2, phosphatidylethanolamine binding protein 1 pseudogene 2; other abbreviations are shown in Figures 1–3.
SMC proliferation after vascular damage is essential for neointimal vessel remodeling. Mounting evidence suggests that histone acetylation is a significant epigenetic alteration in the transcriptional regulation of proliferation-related genes. HDAC, a key regulator of transcriptional cascades, modulates SMC proliferation. A novel epigenetic mechanism reveal that the lysine methyltransferase SMYD2 is overexpressed during injury-induced neointima formation and that SMYD2 promotes the expression of HDAC3 through H3K36 trimethylation at its promoter. HDAC3 directly interacts with and deacetylates the serum response factor (SRF), promoting VSMC phenotypic switching and neointimal hyperplasia (Zhong et al., 2024). Furthermore, inhibiting HDAC activity reduces cyclin D1 and neointima formation, thereby preventing proliferative remodeling caused by intravascular injury, which has important implications for vascular proliferative diseases such as AS (Findeisen et al., 2011).
NcRNAs are also important for VSMC proliferation. miR-143 and miR-145, two of the most abundantly expressed miRNAs in VSMCs under normal conditions, are downregulated in damaged and atherosclerotic regions. Decreased levels of miR-143 and miR-145 may facilitate VSMC proliferation by directly targeting the transcription of SRF. Restoring these miRNAs can prevent smooth muscle hyperplasia associated with vascular damage and AS (Cordes et al., 2009; Elia et al., 2009). CARMN is a lncRNA located upstream of miR-143 and miR-145. CARMN knockout inhibits the expression of miR-143 and miR-145 under normal conditions. Nevertheless, as a microRNA host gene, it has miRNA-independent effects on SMC function. Loss of CARMN controls the functional switch of SMCs towards a pro-atherogenic phenotype and accelerates the development of AS (Vacante et al., 2021). A novel lncRNA, PEBP1P2, suppresses proliferation, migration, and phenotypic switching of VSMCs by interacting with cyclin-dependent kinase (CDK) 9. However, reduced PEBP1P2 levels in the serum of CAD patients indicate a loss of function (He et al., 2020). In addition, circRNAs can regulate gene expression by interacting with specific miRNAs. CircMAP3K5 has been shown to inhibit SMC proliferation in injured arteries by acting as a TET2-mediated competing endogenous miR-22-3p sponge, leading to reduced neointimal formation. Targeting the circMAP3K5/miR-22-3p/TET2 axis may offer a potential therapeutic strategy for AS (Zeng et al., 2021).
3.4 BiomarkersVarious epigenetic changes have been identified in multiple cells implicated in the pathogenesis of AS. However, certain molecules that carry epigenetic information have also been detected in the extracellular interstitial space and body fluids. These molecules are predominantly released into body fluids by diseased tissue cells, exhibit functional activity relevant to the disease, and have the potential to serve as promising biomarkers. Numerous epigenetic biomarkers are associated with CAD and its progression, among which potential non-coding RNA biomarkers are listed in Table 1.
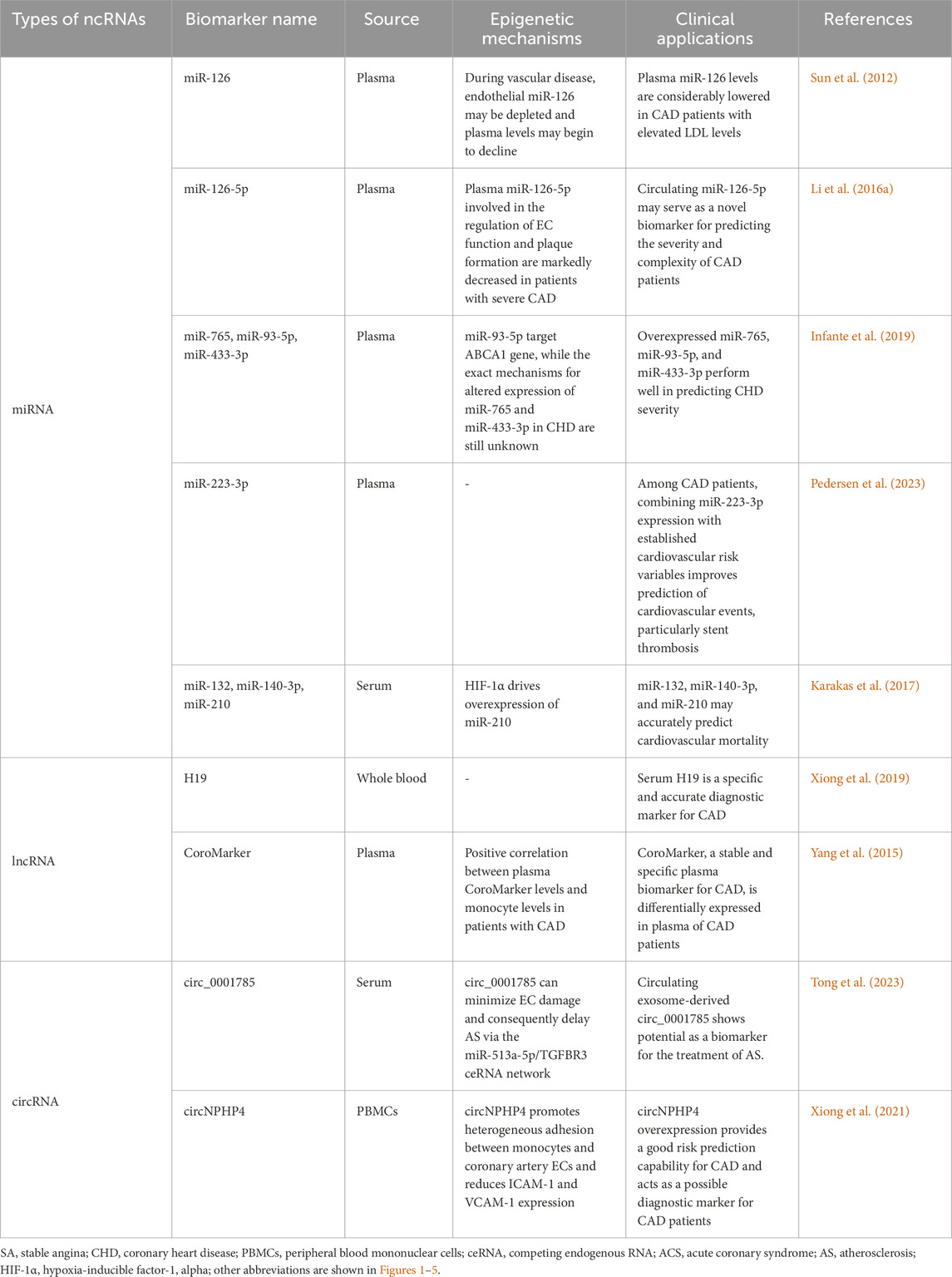
Table 1. Summary of potential non-coding RNAs as biomarkers in CAD.
The DNA methylation patterns of specific genes related to AS have been proposed as biomarkers of CAD. The DNA methylation patterns of tissue inhibitors of metalloprotease 1 (TIMP1), ABCA1, and acetyl-CoA acetyltransferase 1 (ACAT1) are considerably altered in AS patients and function as biomarkers for the early identification of AS (Ma et al., 2016). Another study analyzed DNA methylation changes on specimens of blood from patients with AS, and differentially methylated regions of cysteine-rich secretory protein 2 (CRISP2) and breast cancer 1 (BRCA1), which regulate endothelial function and angiogenesis, respectively, were found to be the most important DNA methylation biomarkers (Istas et al., 2017). Moreover, cell plasticity in vulnerable plaques is associated with tissue-specific alterations in the dynamic DNA methylation patterns. Since atherosclerotic plaques likely release DNA fragments that carry plaque-specific methylation marks into the circulation, researchers have recently proposed that cell-free DNA methylation (cfDNAme) acts as a non-intrusive and tissue-specific biomarker to provide accurate and rapid information about the condition of atherosclerotic plaques and direct patient risk assessment (Diez Benavente et al., 2024). The first applications of cfDNAme have been reported in cardiovascular diseases and may be applied to CAD patients in the future (De Borre et al., 2023).
In addition, several ncRNAs have been detected in extracellular fluids and circulation. Circulating ncRNAs are remarkably stable and can attach to many types of carrier proteins and exosomes, making them easily detectable. Consequently, ncRNAs may act as important biomarkers for CAD and could potentially serve as new therapeutic targets (de Gonzalo-Calvo et al., 2017; Garcia-Gimenez et al., 2017). miR-126 is a potential biomarker of CAD, with its expression influenced by various CAD risk factors, including aging and dyslipidemia (Fichtlscherer et al., 2010; Sun et al., 2012). In particular, the downregulation of circulating miR-126-5p levels is associated with the severity of CAD. miR-126-5p levels are markedly decreased in CAD patients with multi-vessel disease (Li H. et al., 2016). Similarly, the adjusted multivariate regression model reveals that circulating levels of miR-93-5p, miR-433-3p, and miR-765 are increased in patients with critical coronary stenosis, with a good performance in predicting the severity of CAD. The specific receiver operating characteristic (ROC) curve analysis shows that the areas under the curve (AUC) of circulating miR-765, miR-93-5p, and miR-433-3p expression are 0.762 (p < 0.001), 0.770 (p < 0.001) and 0.771 (p < 0.001), respectively (Infante et al., 2019). Moreover, a recent study suggested that adding miR-223-3p expression to established cardiovascular risk variables such as age, gender, smoking, and diabetes mellitus could enhance the prediction of cardiovascular events in individuals with stable CAD, particularly increasing the predictive value of stent thrombosis (AUC: 0.88 vs. 0.77, p = 0.04) (Pedersen et al., 2023). Individual miRNAs extracted from peripheral blood, particularly miR-210 (AUC: 0.754), miR-140-3p (AUC: 0.756), and miR-132 (AUC: 0.737), can precisely predict mortality in patients with CAD as valuable biomarkers for risk estimation (Karakas et al., 2017). Subsequently, research on lncRNAs in CAD has identified several lncRNAs that could be used as biomarkers for diagnostic and prognostic (Li H. Y. et al., 2016; Arslan et al., 2017). Circulating H19 lncRNA levels in CAD patients are significantly elevated compared with healthy individuals with AUC up to 0.9367 (p < 0.001), making it a potential diagnostic biomarker for CAD (Xiong et al., 2019). Additionally, a novel lncRNA CoroMarker, mainly found in EVs, is a stable, accurate, and unique biomarker of CAD. By constructing a diagnostic model with Fisher’s criteria, the sensitivity and specificity of diagnostic accuracy for CoroMarker alone are 68.29% and 91.89%, respectively (Yang et al., 2015). CircRNAs also are stable and easily detectable, emerging as essential diagnostic tools (Li et al., 2018). Exosome-derived circ_0001785, a novel biomarker of atherogenesis, is reduced in the circulating peripheral blood of CAD patients. It is proved in vivo to protect ECs from damage, thereby delaying AS (Tong et al., 2023). Small EV-derived circNPHP4 is significantly upregulated in CAD-related monocytes. ROC analysis performed with circNPHP4 shows high diagnostic performance, as reflected by the Youden index (sensitivity, 87.1%; specificity, 69.7%) (Xiong et al., 2021). These studies highlight the diagnostic and predictive potential of ncRNAs, emphasizing their growing role as prospective biomarkers for CAD. Together, we summarize the clinical applications of these epigenetic biomarkers described above in Figure 5.

Figure 5. Flowchart of potential clinical applications of epigenetic biomarkers in CAD. This flowchart illustrates the application of representative biomarkers in the diagnosis of CAD, prediction of CAD severity, prediction of cardiovascular events in CAD, and prediction of mortality in CAD. TIMP1, tissue inhibitors of metalloprotease 1; ACAT1, acetyl-CoA acetyltransferase 1; DMR, differentially methylated region; CRISP2, cysteine-rich secretory protein 2; BRCA1, breast cancer 1; other abbreviations are shown in Figures 1–4.
To detect these markers, liquid biopsy combined with new molecular analysis techniques is the most promising approach. For cardiovascular diseases, such as CAD, whose affected tissues are not easily accessible, liquid biopsy offers a simple, non-invasive, and reproducible alternative for obtaining dynamic molecular information reflecting the disease state. Typically, liquid biopsies are predominantly performed using blood and are used to analyze a range of biomarkers present in body fluids (Nikanjam et al., 2022; Sharma et al., 2024). As previously mentioned, these CAD-related biomarkers include free extracellular ncRNAs and DNA fragments carrying methylation markers released into the circulation by atherosclerotic plaques. Furthermore, the development of molecular biology techniques, such as polymerase chain reaction (PCR), microarray technology, and high-throughput sequencing, has enabled the acquisition of dynamic molecular information on the state of disease (Xu et al., 2011; Saliminejad et al., 2019; Kojabad et al., 2021). Consequently, in the future, a combination of traditional biomarkers with these newly discovered molecules should be considered in a multi-marker strategy. This could help to monitor disease conditions more accurately, significantly enhance cardiovascular risk management, and guide its treatment.
4 Epigenetic treatment for CADEpigenetics plays a critical role in CAD by regulating various biological processes at multiple levels, from pathogenesis to potential therapeutic interventions. DNA methyltransferase inhibitors (DNMTi), histone deacetylase inhibitors (HDACi), bromodomain and extra-terminal motif inhibitors (BETi), enhancer of zeste homolog 2 EZH2 inhibitors (EZH2i), and ncRNA-based regulation have been widely studied as potential therapeutics for CAD. Furthermore, the specific functions of several chemicals used in clinical practice can be explained by epigenetic pathways. Table 2 provides an overview of specific drugs targeting epigenetic mechanisms (epidrugs). Epigenetic therapies related to gene regulation may offer novel potential strategies to manage CAD, some of which are already being applied in clinical settings.
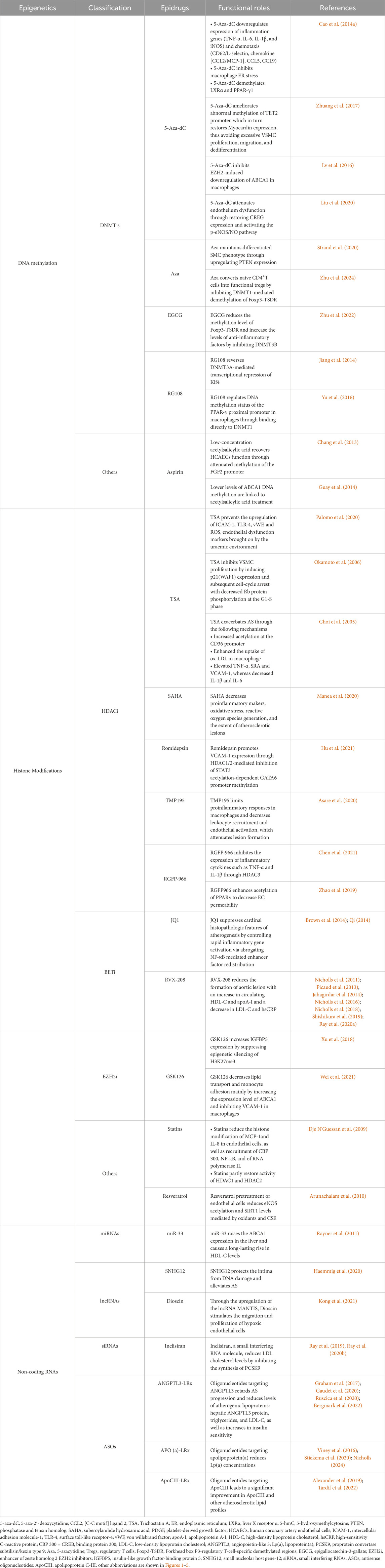
Table 2. Potential epidrugs for the treatment of CAD.
4.1 DNA methylationChanges in DNA methylation are reversible, providing promising prospects for disease treatment. Research on DNA methylation-based treatments for cardiovascular diseases remains in its early stages. Future studies should focus on identifying potential epidrug targets such as DNMT inhibitors. DNMT inhibitors and similar medications help treat CAD by modulating the methylation and expression of target genes.
5-Aza-2′-deoxycytidine (5-Aza-dC), a DNA methyltransferase inhibitor also known as decitabine, is an anticancer drug that significantly attenuates atherosclerotic lesions. 5-Aza-dC reduces the infiltration of macrophages in AS, which is correlated to decreased expression of inflammatory and chemotaxis genes, as well as suppressed macrophage endoplasmic reticulum stress. Additionally, 5-aza-dC demethylates PPAR-γ1 and liver X receptor α (LXRα) promoters that lead to overexpression of LXRα and PPAR-γ (Cao et al., 2014b). It simultaneously reduces the global 5-methylcytosine content and restores myocardin expression in VSMCs stimulated by platelet-derived growth factors (PDGF), thereby limiting excessive VSMC dedifferentiation and vascular remodeling (Zhuang et al., 2017). Moreover, 5-aza-dC prevents the downregulation of ABCA1 expression (Lv et al., 2016). Recently, it has been demonstrated that 5-aza-dC substantially attenuates ox-LDL-induced EC dysfunction by rescuing CREG expression (Liu et al., 2020). Overall, these mechanisms contribute to 5-aza-dC’s protective effect. Other researchers have also explored the potential of the demethylating drug 5-azacytidine (Aza) as therapy for AS. Aza treatment blocks PDGF-induced SMC dedifferentiation via phosphatase and tensin homolog (PTEN) upregulation, a crucial regulator of the SMC phenotype (Strand et al., 2020). Besides, CD4+T cells are induced by Aza in vitro to generate induced regulatory T cells (iTregs), adoptive transfer of which attenuates AS due to increasing peripheral blood Tregs and suppressing inflammation. As a demethylating drug, Aza converts naive CD4+T cells into functional Tregs by inhibiting DNMT1-mediated demethylation of Forkhead box P3 (Foxp3)-regulatory T-cell-specific demethylated regions (TSDR) (Zhu et al., 2024). Similarly, epigallocatechin-3-gallate (EGCG), a green tea-derived phenol, also leads to a reduction in Foxp3-TSDR methylation levels, which is achieved through the inhibition of DNMT3B. Meanwhile, a notable increase in peripheral blood treg levels is observed, along with elevated transforming growth factor-beta (TGF-β) and IL-10, and a decrease in IL-β and Interferon-gamma (IFN-γ) levels, thereby attenuating atherosclerosis (Zhu et al., 2022).
Different from the above DNMT inhibitors, RG108 is a non-nucleoside analog, showing an inhibitory effect toward DNMT3A. Hemodynamic disturbed flow is considered to be associated with atherosclerosis susceptibility by inducing DNA methylation of the Klf4 promoter in ECs. RG108 reverses this DNMT3A-mediated transcriptional repression of Klf4 and restores its anti-inflammatory protective effects (Jiang et al., 2014). Furthermore, it has been demonstrated that this compound is also capable of binding directly to DNMT1, thereby inhibiting its activity. DNMT1 has been shown to regulate the DNA methylation status of the PPAR-γ proximal promoter in macrophages during the development of AS (Yu et al., 2016). Inhibition of DNMT1 by RG108 could be considered a potential strategy for the treatment of CAD.
Furthermore, there is evidence that certain pharmaceuticals can exert therapeutic effects by modulating the methylation status of genes. For example, aspirin, a standard medication under current rules, has been shown to affect gene methylation. L5, the sole subfraction of LDL, which causes apoptosis in cultured ECs by increasing methylation of the fibroblast growth factor-2 (FGF2) promoter, is significantly increased in patients with CAD. Low-dose aspirin attenuates the detrimental effect of L5 by attenuating this methylation effect (Chang et al., 2013). Aspirin therapy also lowers ABCA1 DNA methylation levels, thereby stimulating reverse cholesterol transport and reducing the incidence of AS and CAD (Guay et al., 2014).
To summarize, a variety of DNMT inhibitors have been extensively studied in the context of CAD, with promising results indicating their potential for treatment. Further in-depth studies are recommended in the future to determine the efficacy of drugs based on DNA methylation modulation in benefitting patients with CAD.
4.2 Histone modificationsAmong the multiple histone modifications, histone acetylation and methylation are crucial in treating CAD. Key enzymes and functional proteins within these modification processes can be used as therapeutic targets for managing CAD.
4.2.1 HDACiTrichostatin A (TSA), a potent inhibitor of HDAC, has been extensively researched in the pathogenesis of CAD. TSA prevents the upregulation of EC dysfunction markers induced by the uremic milieu, such as intercellular adhesion molecule-1, von Willebrand Factor, and ROS, whereas EC dysfunction is an early cause of AS in chronic kidney disease (CKD). Therefore, we hypothesized that a specific blockade of HDAC by TSA would prevent CKD-induced AS (Palomo et al., 2020). In addition, TSA inhibits VSMC proliferation by inducing cell cycle arrest, which may attenuate the progression of AS (Okamoto et al., 2006). As a result, TSA has been considered for treating AS and CAD. Nevertheless, the first mouse study on AS treatment with TSA found that it enhanced atherosclerotic lesion formation. TSA increased the acetylation of the CD36 promoter region in macrophages, enhancing the intake of oxLDL. Furthermore, scavenger receptor A (SRA), tumor necrosis factor-alpha (TNF-α), and VCAM-1 were also elevated with TSA treatment, exacerbating AS (Choi et al., 2005). These results show that elevated histone acetylation can affect the progression of AS by altering the expression of ox-LDL receptors and proatherogenic genes. In addition to TSA, treatment with suberoylanilide hydroxamic acid (SAHA), a pan-HDAC inhibitor proved to be one of the antineoplastic drugs, decreased the extent of atherosclerotic lesions in ApoE−/− mice, possibly through a combination of mechanisms that included negative regulation of oxidative stress and proinflammatory marker expression (Manea et al., 2020). Moreover, Romidepsin is another approved HDACi with a unique structure, and it reduces diet-induced atherosclerotic lesion accumulation in Apoe−/−mice through simultaneous inhibition of HDAC1/2 (Hu et al., 2021). In brief, these data suggest that pharmaceutical treatment focused on HDACs is an effective therapeutic approach for AS.
Further studies have shown the role of targeting specific HDACs in AS. Deletion of HDAC9 reduces AS in vivo (Cao et al., 2014a). Concurrently, lesion formation was attenuated by blocking HDAC9 with the class IIa HDAC inhibitor TMP195. This blockade limited proinflammatory responses in macrophages and decreased endothelial activation and leukocyte recruitment. Moreover, TMP195 diminishes plaque vulnerability, improving plaque stability in advanced lesions. Monocytes from individuals with established AS produced fewer inflammatory cytokines, including IL-1β and IL-6, when treated ex vivo (Asare et al., 2020). Besides, HDAC3 also represents a highly effective target, and the development of small-molecule inhibitors that specifically target this protein is currently a major area of research. HDAC3-selective inhibitor RGFP-966 alleviates atherosclerotic lesions and inhibits endothelial-to-mesenchymal transition in atherosclerotic plaques. In vitro studies show that RGFP-966 inhibits the expression of inflammatory cytokines such as TNF-α and IL-1β through HDAC3, thereby suppressing the inflammatory response of ECs (Chen et al., 2021). The inhibition of HDAC3 with RGFP966
Comments (0)