Cell lines
HEK293T (a female cell line, ATCC: CRL-3216) was purchased from ATCC and kindly provided by Dr. Junjie Chen (MD Anderson Cancer Center). HEK293A (a female cell line, Thermo Fisher Scientific: R70507) cells were kindly provided by Dr. Jae-Il Park (MD Anderson Cancer Center). HEY (a female cell line, ATCC: CRL-3252), HEC1A (a female cell line, ATCC: HTB-112), and CAL-27 (a male cell line, ATCC: CRL-2095) cell lines were purchased from ATCC. HEK293T, HEK293A and CAL-27 cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM). HEY cells were maintained in RPMI-1640 Medium. HEC1A cells were maintained in McCoy's 5A Medium. All the culture media were supplemented with 10% fetal bovine serum and contain 1% penicillin and streptomycin. All the cells were maintained at 37 °C in 5% CO2 (v/v).
Antibodies and chemicals
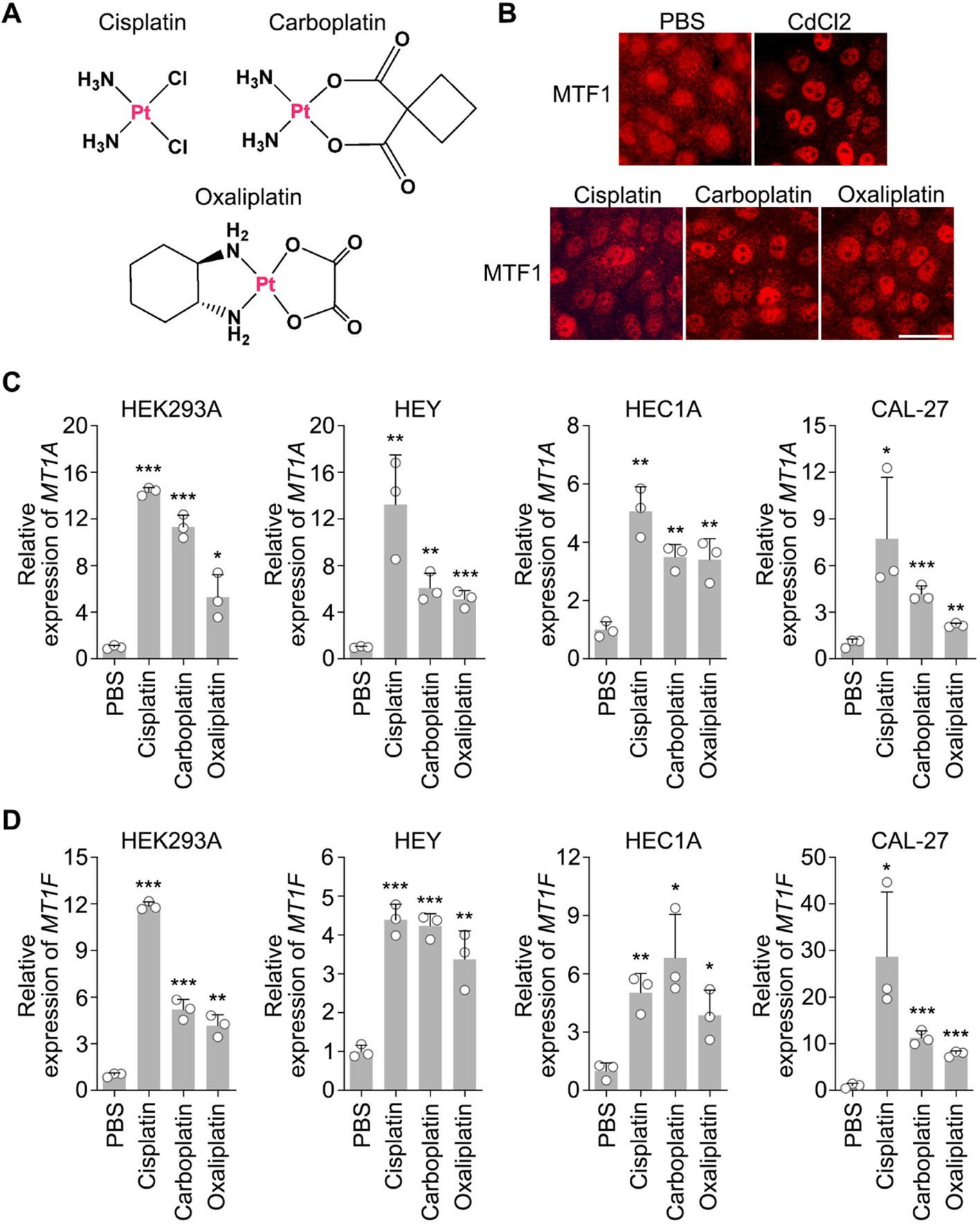
For Western blotting, anti-MTF1 (NBP1-86,380, 1:2000 dilution) antibody was obtained from Novus Biologicals. Anti-Flag (M2)-peroxidase (HRP) (A8592-1MG, 1:5000 dilution) and anti-α-tubulin (T6199-200UL, 1:5000 dilution) antibodies were obtained from Sigma-Aldrich. Anti-phospho-LATS1 (Thr1079) (8654S, 1:1000 dilution) and anti-LATS1 (3477S, 1:1000 dilution) antibodies were purchased from Cell Signaling Technology. For immunofluorescent staining, anti-MTF1 antibody (NBP1-86,380, 1:200 dilution) was obtained from Novus Biologicals.
Cisplatin (AG-CR1-3590) was purchased from Adipogen. Carboplatin (C2538), oxaliplatin (O9512), FIPI (F5870), cerivastatin (SML0005), and CdCl2 (655,198) were purchased from Sigma-Aldrich. Mitomycin C (HY-13316) and camptothecin (HY-16560) were obtained from Med Chem Express.
Constructs and viruses
Plasmids encoding the indicated genes were obtained from the Human ORFeome V5.1 library or purchased from DNASU Plasmid Repository. The constructs were generated via polymerase chain reaction (PCR) and sub-cloned into a pDONOR201 vector using Gateway Technology (Thermo Fisher Scientific) as entry plasmids. A lentiviral gateway-compatible destination vector with the SFB tag was used to express various fusion proteins. PCR-based mutagenesis was used to generate the indicated site mutations.
Lentiviral supernatants were generated by transient transfection of HEK293T cells with the helper plasmids pSPAX2 and pMD2G and harvested 48 h later. Supernatants were passed through a 0.45-µm filter and used to infect cells with the addition of 8 µg/mL hexadimethrine bromide (Polybrene) (Sigma-Aldrich). Plasmid transfection was performed using polyethyleneimine (PEI) (23,966–2, Polysciences).
Bioinformatic analysis
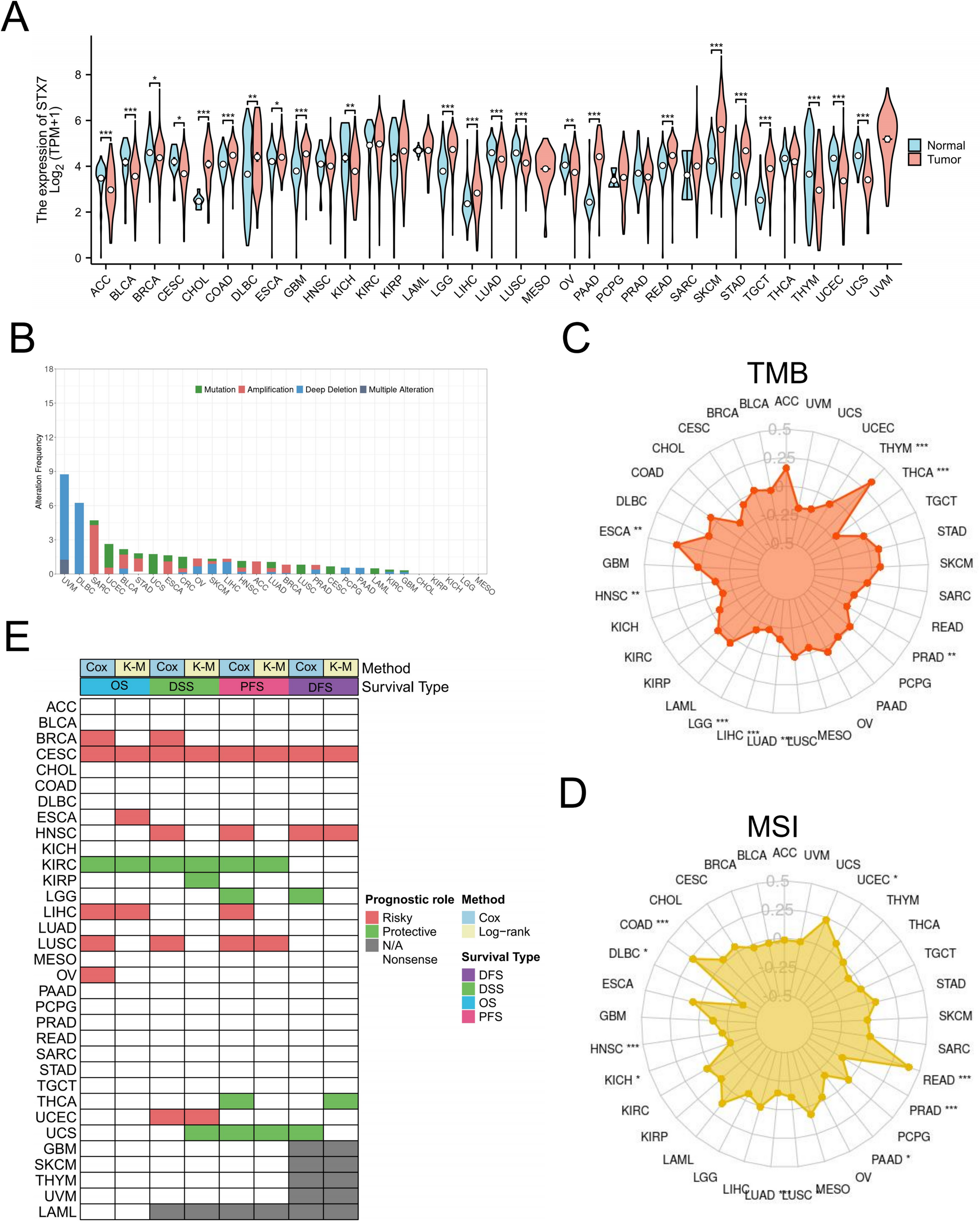
Clinical information of genes CTGF, CYR61, MT1A, MT2A, YAP1, WWTR1 and MTF1 was obtained from The Cancer Genome Atlas (TCGA) data portal (http://tcga-data.nci.nih.gov). mRNA expression level of the indicated genes was analyzed in lung cancer (TCGA-LUAD) and head and neck cancer (TCGA-HNSC) using GraphPad spearman correlation. Each point indicates an individual tissue sample. Additionally, mRNA expression level of MT1-family genes and MT2A was analyzed in breast and ovarian cancers using Kaplan–Meier Plotter (https://kmplot.com/analysis/). R, correlation coefficient. As for the patient survival studies, the top one-third expression level of each gene among all patients was classified as high, while the remaining two-thirds were classified as low. Survival data for each group were analyzed by log-rank (Mantel-Cox) test.
Generation of MTF1 knockout (KO) cell lines using CRISPR/Cas9
The indicated LATS1/2 double knockout (DKO), MOB1A/B DKO, NF2 KO, MTF1 KO, LATS1/2/MTF1 triple knockout (TKO) HEK293A cells were generated as described previously [25, 27]. The same single-guide RNAs (sgRNA) set was used to generate the MTF1 KO HEY, HEC1A and CAL-27 cells. Briefly, five sgRNAs of MTF1 were designed by CHOPCHOP website (https://chopchop.rc.fas.harvard.edu), cloned into the lentiGuide-Puro vector (Addgene plasmid # 52,963), and transfected into each cancer cell line with the lentiCas9-Blast construct (Addgene plasmid # 52,962). The next day, cells were selected with puromycin (2µg/ml) for two days and subcloned to form single colonies. Knockout cell clones were screened by Western blot to verify the loss of MTF1 expression. The genomic editing was further confirmed by sequencing.
Immunofluorescent staining
Immunofluorescent staining was performed as described previously [28]. Briefly, cells cultured on coverslips were fixed with 4% paraformaldehyde for 10 min at room temperature and then extracted with 0.5% Triton X-100 solution for 5 min. After blocking with Tris-buffered saline with Tween 20 containing 1% bovine serum albumin, the cells were incubated with the indicated primary antibodies for 1 h at room temperature. After that, cells were washed and incubated with rhodamine-conjugated secondary antibodies for 1 h. To visualize nuclear DNA, cells were counterstained with 100 ng/mL 4′,6-diamidino-2-phenylindole (DAPI) for 2 min. The cover slips were mounted onto glass slides with an anti-fade solution and visualized under a Nikon Ti2-E inverted microscope.
Cell viability assay
For crystal violet staining assay, cells were seeded in 12-well plates and subjected to the indicated treatments. At the endpoint, cells were fixed with 4% paraformaldehyde for 10 min and stained with 0.1% crystal violet. Cells were then washed for three times and detained with acetic acid. The absorbance of the crystal violet solution was measured at O.D. 595 nm and normalized to vehicle-treated cells.
RNA extraction, reverse transcription, and real-time PCR
RNA samples were extracted with TRIzol reagent (Invitrogen). Reverse transcription assay was performed with the Script Reverse Transcription Supermix Kit (Bio-Rad) according to the manufacturer’s instruction. Real-time PCR was performed using Power SYBR Green PCR master mix (Applied Biosystems). For quantification of gene expression, the 2−ΔΔCt method was used. GAPDH expression was used for normalization. The sequence information of q-PCR primers used for gene expression analysis is as follows:
MT1A-Forward: 5′- CAGCTGCACTTCTCTGATGC-3′;
MT1A-Reverse: 5′- AATGCAACTCCTGCAAGAAGA-3′;
MT1F-Forward: 5′- TCTCTTGGAAAGTCCAGTCTC-3′;
MT1F-Reverse: 5′- ACTCTTTGCACTTGCAGGA-3′;
GAPDH-Forward: 5′-ATGGGGAAGGTGAAGGTCG-3′;
GAPDH-Reverse: 5′-GGGGTCATTGATGGCAACAATA-3′.
Xenograft tumor assays
All the xenograft tumor assays were followed with institutional guidelines, approved by the Institutional Animal Care and Use Committee (IACUC; protocol number AUP-19–112) of University of California, Irvine (UCI), and performed under veterinary supervision. Athymic nude (nu/nu) mouse strain was used for the xenograft tumor assays in this study. All the nude mice were purchased from Jackson Laboratory and kept in a pathogen-free environment at the UCI ULAR facility. The indicated HEY and HEC1A cells (2 × 106) were subcutaneously injected into nude mice. When tumors were approximately 50 mm3 in size, 10 mice for each cell line were randomly assigned into two groups (5 mice per group) and subjected to cisplatin treatment (5 mg/kg) twice a week. After three weeks for treatment, mice were euthanized, and tumor weights were analyzed.
For subcutaneous injection, isoflurane anesthesia was used to release the pain of mice. Specifically, mice were anesthetized with an initial concentration of 4‐5% isoflurane mixed with oxygen in an induction chamber. When mice were unconscious, they were transferred to a nosecone to continue delivery of isoflurane in oxygen. Depth of anesthesia was evaluated by lack of response to toe pinch, touching of eyelids or other stimulus, and evaluation of the respiration rate. Anesthesia depth was re‐assessed at 2‐3 min intervals for the duration of the procedure. Consistent with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association, all animals were sacrificed by means of inhalation of carbon dioxide gas. Carbon dioxide was introduced into the cage at a 10 ~ 30% fill rate per minute until no movement of mice was noted for at least one minute. Mice were subsequently subjected to cervical dislocation as a secondary method to verify euthanasia.
Quantification and statistical analysis
Patient survival data were analyzed by log-rank (i.e., Mantel-Cox) test. Gene correlation data were analyzed by spearman correlation. There were no samples or animals excluded from studies. There was no statistical method used to predetermine sample size for the mouse xenograft tumor experiments. The Student’s t-test (two-sided) was used to analyze the differences between two sample groups. SD was used for error estimation. Each experiment was repeated twice or more, unless otherwise noted. A p value < 0.05 was considered statistically significant.


Comments (0)