Biochemical and molecular analyses are essential to identify cases of AFD, but sometimes the results can be inconclusive, prompting the use of histological evaluations to help the diagnosis. Kidney biopsy is a cornerstone in nephrology for diagnosing renal diseases and guiding treatment decisions. With advancements in radiological guidance, renal biopsy has become a safe and routine procedure [6]. Although genetic testing is widely available for the diagnosis of AFD, kidney biopsy remains a valuable tool, especially in complex genetic cases, such as variants of unknown significance (VUS) [7] or non-coding GLA variants that are not routinely included in standard diagnostic protocols. Recent studies [8, 9] have examined the actual pathogenic effects of non-coding mutations, demonstrating that they can have an impact similar to pathogenic variants occurring in coding regions. These mutations may directly alter the enzyme’s peptide sequence, structure, and function, thereby contributing to disease pathogenesis. However, even in these cases the utility of kidney biopsies continues to be a subject of debate [10].
Histological findings, as widely described in the literature, provide valuable insights into the severity of renal involvement, often revealing subclinical damage and response to treatment [11, 12]. Studies by the International Study Group of Fabry Nephropathy (ISGFN) underscore the utility of kidney biopsy as part of the baseline assessment of the AFD nephropathy [13]. The real chronicity status pointed out by glomerulosclerosis, interstitial fibrosis and arteriolar hyalinosis, was detected even in patients at incipient stages with minimal or absent proteinuria. These findings suggest that histological renal involvement precedes overt clinical signs, highlighting the value of early detection.
All observed structures exhibit lipid inclusions and show early stages of sclerosis, even in patients with non-classical mutations and VUS [14]. Fibrosis, as observed in other chronic diseases, represents the final stage of tissue damage; however, few studies have investigated the pathogenesis of fibrosis in AFD nephropathy. The deposition of Gb3 in various renal cell types triggers biological processes that induce the production of pro-inflammatory cytokines and promote fibroblast proliferation [15]. At present, kidney biopsy remains the only reliable method to assess the degree of kidney fibrosis, unlike other organs such as the heart and brain, where innovative imaging techniques provide sensitive evaluations of fibrosis. It is well-established that fibrosis can limit the therapeutic response to ERT. Therefore, a deeper understanding of the molecular mechanisms underlying fibrosis may lead to the development of new therapeutic strategies [16]. Additionally, confocal microscopy studies using fluorescent mouse monoclonal anti-Gb3 antibodies can be applied to biopsy specimens to confirm Gb3 deposits in ambiguous cases [17].
Regarding the prognostic value of kidney biopsy, recent studies by Michael Mauer and Behzad Najafian have focused on podocyte involvement in the progression of AFD nephropathy. By leveraging ultrastructural morphometric measurements of Gb3 inclusions, their research developed a quantification method to describe the percentage of podocytes exhibiting the AFD phenotype. This method is adjusted for age and sex while addressing the heterogeneity of podocyte involvement in female mosacism [18,19,20]. Future research goals include establishing a rigorous histology-based risk stratification model for disease progression and identifying a hypothetical threshold for podocyte Gb3 inclusion volume to mitigate glomerular stress through early treatment.
Lastly, kidney biopsy provides distinctive features that can aid in differentiating superimposed renal diseases, especially when the clinical context, genetic data, and family history are unclear [21].
The two cases presented here illustrate the role of kidney biopsy in resolving diagnostic challenges in AFD and demonstrate its critical role when clinical and laboratory findings alone are insufficient for a definitive diagnosis. Our experience emphasizes the significance of kidney biopsy in cases with complex genetic backgrounds (patient 1), while highlighting the importance of a multidisciplinary approach in addressing the diagnostic challenges posed by overlapping histological features (patient 2).
In the first case, kidney biopsy played a pivotal role in clarifying the diagnosis despite discrepancies between clinical and laboratory findings. The histological suspicion prompted further diagnostic evaluation, including genetic analysis of fibroblast cultures, which ultimately led to a precise diagnosis of AFD. A key lesson from this case is that in patients with a clinical phenotype consistent with AFD, but no identified GLA gene variant, it is important to thoroughly review genotyping data and consider alternative molecular diagnostic methods.
On the other hand, the second case highlights the limitations of relying solely on histological findings for diagnosis and underscores the importance of a multidisciplinary approach that integrates genetic testing, second level histological analysis, and comprehensive clinical and laboratory evaluations to rule out misleading diagnoses. In this patient, the pathology report alone was confusing and confirmed the value of enzymatic and genetic testing.
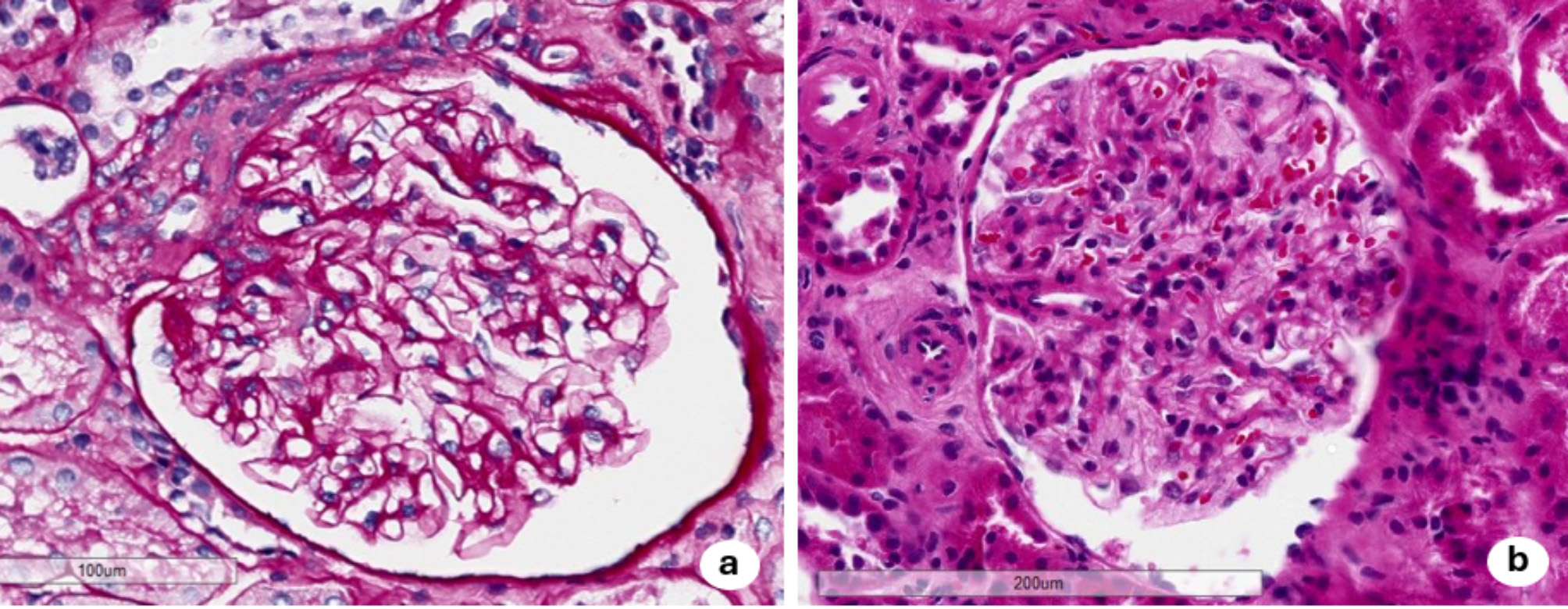
Histological features should always be interpreted in conjunction with clinical data in a multidisciplinary setting. Myeloid bodies, indeed, are not an uncommon as ultrastructural finding in kidney biopsies, and they can also be found in patients without clinical evidence of AFD [22, 23]. Therefore, further work-up is necessary to exclude drug- induced phospholipidosis (DIP), toxins, and other inherited diseases such as Nail Patella Syndrome or Niemann-Pick disease [24]. Despite overlapping histological features, distinguishing between AFD and these other conditions is possible. In non-AFD kidney biopsies, myeloid bodies are focal and predominantly localized within podocytes. Furthermore, the inclusions tend to be smaller, scattered, and may completely resolve upon withdrawal of the inciting agent. In contrast, AFD kidney biopsies exhibit permanent lamellar inclusions, generally known as Zebra bodies, present in at least two distinct cell types: podocytes, parietal cells, distal tubular epithelium, and endothelial cells [24]. To better differentiate between these two entities, we propose that Zebra bodies should be considered highly diagnostic of AFD, whereas the presence of nonspecific Myeloid bodies requires further context to assess their clinical relevance.
This analysis confirms the importance of a histological evaluation conducted by expert pathologists, in collaboration with clinical teams, to accurately characterize the nature of deposits and resolve diagnostic dilemmas. In rare diseases like AFD, an integrated, multidisciplinary approach is crucial for achieving an accurate diagnosis and avoiding misdiagnosis or delays. Podocyte vacuolization and intracellular lamellar inclusions observed on electron microscopy, in the context of other characteristic features, may confirm the diagnosis of this lysosomal storage disorder, but they can also be secondary to other conditions. Following these considerations, the essential role of histological evaluation clearly comes into light as both a decisive tool and a potential source of diagnostic confusion in the complex clinical setting of AFD. Prompt recognition of these challenges and the implementation of a comprehensive evaluation will help ensure optimal patient outcomes.




Comments (0)