
Remember me


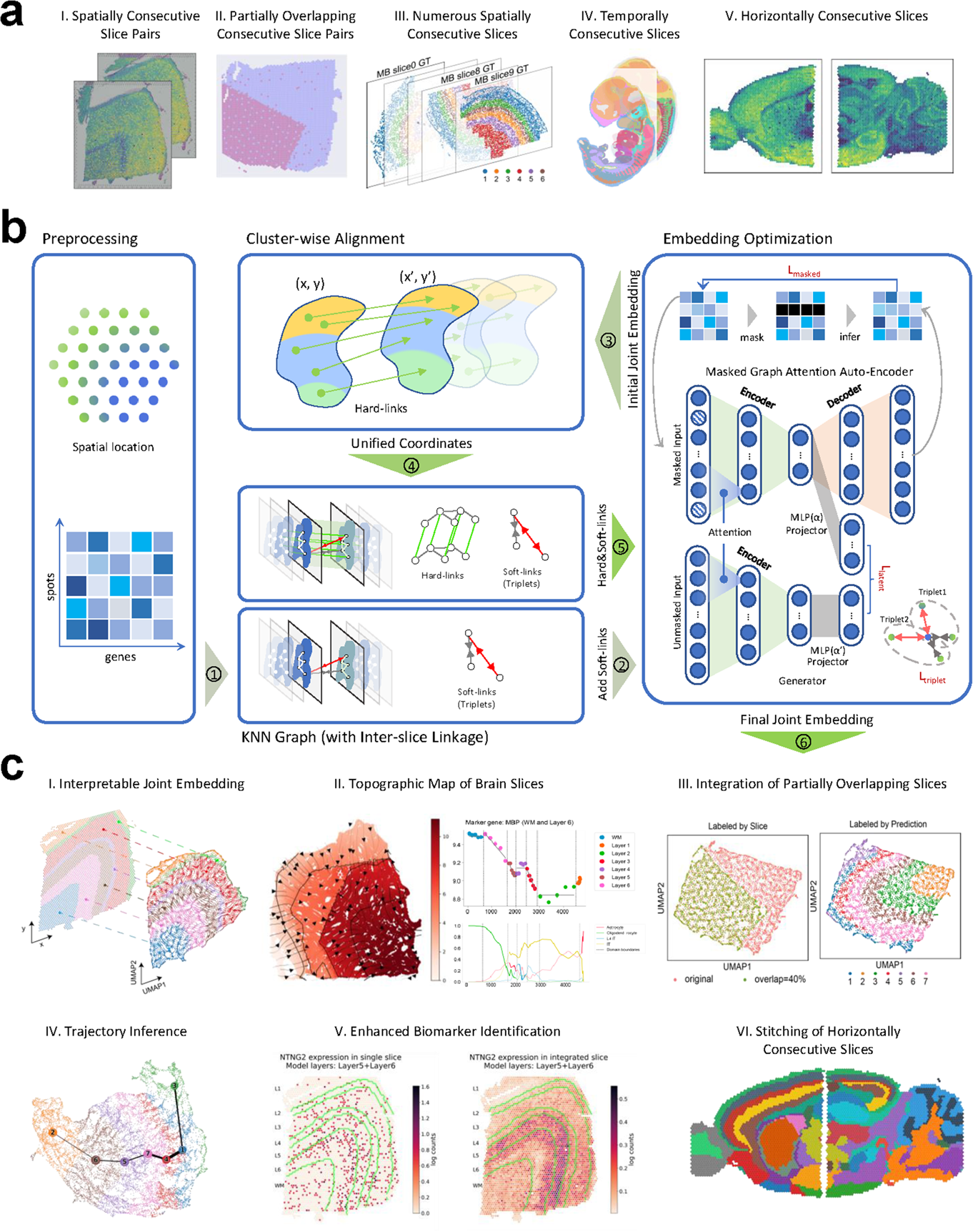




Capturing the full gene-splicing events, particularly for those occurring with low abundance, is critical to reveal the heterogeneity of various gene isoforms from different AS events. To achieve this, we substantially increased the sequencing depth of RNA-seq with an average of 120 M 150-bp long paired-end sequencing reads per sample, which is about 530 × coverage of the AtRTD3 reference transcriptome [40] (Fig. 1A; Additional file 1: Table S1). We first analyzed the RNA-seq datasets from the Arabidopsis ecotype Col-0 seedlings treated with 100 nM flg22 or H2O (mock) for 60 min, which is commonly used to induce PTI in Arabidopsis [20, 41, 42]. No significant batch-to-batch variation from three biological repeats was found using principal component analysis (PCA) with RNA-seq data from total transcripts and genes (Additional file 2: Fig. S1A). The flg22 treatment was validated by well-studied early PTI marker genes, including WRKY30, WRKY33, and FLG22-INDUCED RECEPTOR-LIKE KINASE1 (FRK1) from the RNA-seq data (Additional file 2: Fig. S1B). The trimmed and normalized datasets were subjected to the 3D RNA-seq analysis to identify DE genes, DAS genes, and DTU transcripts (Fig. 1A). The pipeline of 3D RNA-seq analysis incorporates state-of-the-art bioinformatic methods and normalizes the expression with data-driven optimal parameters to improve the analytical accuracy [39]. Out of 40,932 genes from the AtRTD3 reference transcriptome [40], 4260 genes (10.4%), comprising 2741 upregulated and 1519 downregulated genes, were identified as flg22-regulated differentially expressed genes (flg22-DEGs) based on cut-off of |fold change|≥ 2 and false discovery rate [FDR] < 0.01 compared to the mock treatment (Fig. 1B; Additional file 2: Fig. S1C; Additional file 1: Table S2).
Fig. 1
Global profiling of alternative splicing events in response to flg22 treatment. A Scheme of in-depth transcriptome profiling for alternative splicing in response to flg22. Two-week-old plate-grown wild-type (WT) Col-0 and cpl3-3 mutant seedlings treated with H2O (mock) or 100 nM flg22 for 60 min were subjected to RNA isolation and sequencing. To capture the transcripts with low abundance, RNA-seq was performed with Illumina HiSeq 2500 to obtain 120 million reads per sample with the paired-end 150-bp read length, which is about 530 × coverage of Arabidopsis transcriptome. The sequenced reads were aligned to the AtRTD3 reference transcriptome for quantification. The 3D RNA-seq analysis was performed to identify differentially expressed genes (DEGs), differentially alternatively spliced genes (DASs), and transcripts with differential transcript usage (DTUs). The bottom left panel shows an example of DEGs with two differentially expressed transcripts, where changes in abundance between conditions (dH2O and flg22 treatments) are measured by log2 fold change. Total gene expression is represented in the blue line, which is the sum of the expression of all individual transcripts (green and yellow lines). The percentage values denote the expression ratios of individual transcripts relative to the total gene expression. The bottom right panel illustrates examples of DAS and DTU. For a DAS gene, it must have more than one transcript, and changes in expression between individual transcripts (green, yellow, and purple lines) and the total gene expression (blue line) are compared between conditions. The change in percent spliced (ΔPS) is the percentage change in the abundance of a transcript relative to the total gene expression. For a gene to be classified as DAS, at least one transcript has a |ΔPS|≥ 0.1. In DTU analysis, individual transcripts show different expression patterns compared to other transcripts of the same gene. DTU is identified by comparing the change in expression of each transcript to the average expression change of the other transcripts within the same gene. In this example, the transcripts represented with green and yellow lines, but not with the purple line, are DTUs. B Flg22 treatment triggers transcriptional changes in gene expression and alternative splicing in WT plants. The Y-axis indicates the numbers of flg22-triggered DEGs and DAS genes, and DTU transcripts. The flg22-triggered up-/downregulated DEGs were identified based on an absolute value of fold change (|FC|) ≥ 2 and false discovery rate (FDR) < 0.01 between mock and flg22 treatment. The flg22-triggered DASs and flg22-triggered DTUs were selected based on an absolute delta percent spliced (|ΔPS|) ≥ 0.1 and FDR < 0.01 between mock and flg22 treatment. C Volcano plot of flg22-triggered DTUs in WT. Up- and downregulated DTUs in response to flg22 in WT were depicted by a volcano plot. Purple and pale purple dots represent up- and downregulated flg22-DTUs, respectively. The DTUs with non-statistically significant differences were indicated as gray. The Y-axis denotes − log10(FDR), while the X-axis shows ΔPS values. The cut-off lines for FDR = 0.01 and ΔPS = ± 0.1 were indicated as blue and green dashed lines, respectively. D Limited overlapping between flg22-DEGs and flg22-DASs. The Venn diagram between flg22-DEGs (orange circle) and flg22-DASs (pink circle) in WT plants shows the percentages and corresponding gene numbers indicated in each group. E Gene ontology (GO) analysis of flg22-DASs in WT. The statistically enriched gene ontology terms were identified based on the frequency of up-/downregulated flg22-DASs annotated to their frequency in the genome with the cut-off of fold enrichment ≥ 1 and false discovery rate (FDR) < 0.05. F Diagrams of gene structures for three representative flg22-DASs in WT. Solid lines indicate introns, black boxes represent exons, and purple boxes denote alternatively spliced regions. Constitutive, constitutive splicing isoform; IR, intron retention. G Diagrams of protein domains for three representative flg22-DASs in WT. Proteins encoded by constitutive and splicing variant transcripts are designated as β and α forms, respectively. Distinct functional domains with various colored boxes were labeled in the figure. H Relative isoform abundances of three representative flg22-DASs in WT. The isoform usage (IU) was calculated by the percentage abundance of a transcript compared to the total expression of the gene. The blue line represents constitutive splicing transcript (β form), which was defined by a major isoform containing all exons among all splicing variants, and the yellow line represents alternative splicing transcripts (α form), respectively. The expression levels of individual transcripts were retrieved from RNA-seq data. I RT-qPCR analysis of individual splicing variants from three representative flg22-DAS genes. Two-week-old seedlings were treated with or without 100 nM flg22 for 60 min for RT-qPCR analysis with primers specific to each splicing variant. Relative expressions of target transcripts were normalized to UBQ10, and data are shown with mean ± S.D. from three biological repeats (n = 3). Data were analyzed by unpaired two-tailed Student’s t-test between mock- and flg22-treatment. Non-statistically (ns) and statistically significant differences with the corresponding p values were indicated in the figure
To determine splicing variant changes in response to flg22 treatment, we analyzed transcript isoform usage (IU) by calculating the percentage abundance of each transcript relative to the total expression of each gene [40]. Subsequently, we determined the values of delta percentage spliced (ΔPS), which represents the difference in IUs between mock and flg22 treatment. If an IU of any transcript from a gene is significantly changed by flg22 treatment (|ΔPS|≥ 0.1 and FDR < 0.01), we called the gene and transcript DAS and DTU, respectively (Fig. 1A). A total of 642 flg22-regulated DAS genes (flg22-DASs), accounting for 1.6% of the total 40,932 genes from AtRTD3, were identified in WT (Fig. 1B; Additional file 1: Table S3). Their corresponding 980 flg22-regulated DTU transcripts (flg22-DTUs) comprised 527 upregulated and 453 downregulated transcripts (Fig. 1C; Additional file 1: Table S4). Notably, 223 genes were shared between flg22-DEGs and flg22-DASs, representing 5.2% of flg22-DEGs and 34.7% of flg22-DASs (Fig. 1D). This observation suggests that flg22 predominantly regulates distinct sets of genes exhibiting either differential expression or splicing to optimize plant defense mechanisms. The functional consequences of AS are reflected in the changes in isoform ratios [43]. We examined isoform switching (IS) events, where a pair of transcripts reverse their relative abundance between two different conditions, representing one of the most prominent isoform usage changes [39]. Our analysis revealed that 77 flg22-DAS genes, accounting for 12%, exhibited ISs between two flg22-DTU transcripts (Additional file 2: Fig. S1D). Gene ontology (GO) enrichment analysis revealed that among 642 flg22-DASs in WT, there was significant enrichment of terms related to immunity, such as response to the bacterium, regulation of defense response, and immune system process (Fig. 1E; Additional file 1: Table S5). Taken together, our findings demonstrate that flg22 elicitation induces significant alterations in both gene expression and AS patterns. Importantly, each of these processes largely regulates distinct groups of genes, highlighting the complexity and specificity of the plant immune response to flg22.
Given that different transcript isoforms of a gene can encode the same functional protein sequence, the flg22-DTU transcripts were annotated for their gene structure and translated functional domains. Among flg22-DTUs, the transcripts having constitutive splicing events from the majority of individual splicing junctions were defined as the constitutive isoform [44], which was denoted as the beta (β) form here based on DGK5 AS nomenclature (see below). The transcripts that encode different protein sequences from the constitutive ones were denoted as the alpha (α) form. Most, if not all, flg22-DASs only have β and α forms. Occasionally, the transcripts encoding protein sequences different from β and α forms were denoted as the gamma (γ) form. Several well-known immunity-related genes, including SOMATIC EMBRYOGENESIS RECEPTOR-LIKE KINASE 4 (SERK4), ENHANCED DISEASE SUSCEPTIBILITY 5 (EDS5), and CYSTEINE-RICH PROTEIN KINASE 29 (CRK29), were identified among flg22-DASs, with substantial read counts (Fig. 1F and G; Additional file 2: Fig. S1E). The SERK4α has an intron retention (IR) splicing variant at the 7th intron (Fig. 1F), resulting in a premature SERK4α protein after the leucine-rich repeat (LRR) domain (Fig. 1G). Lacking the kinase domain, SERK4α is unlikely to be functional. An IR splicing variant at the first intron of EDS5 leads to a truncated and non-functional EDS5α protein that lacks domains after the first transmembrane domain (Fig. 1F and G). The CRK29α bears an IR at the last intron, leading to a truncated protein with an incomplete kinase domain (Fig. 1F and G). Interestingly, SERK4, EDS5, and CRK29 display IS between two isoforms at 60 min upon flg22 treatment (Fig. 1H). The ratio of their α form transcripts increased, whereas the ratio of the β form transcripts decreased upon PTI activation (Fig. 1H). The production of nonfunctional α form transcripts might be a mechanism for plants to balance the immune response via counteracting the functional β forms. The transcript levels of each isoform obtained from RNA-seq data were confirmed by RT-qPCR with primers specific to each isoform (Fig. 1I; Additional file 2: Fig. S1F and G). Notably, the induction of both α and β transcript isoforms for SERK4, EDS5, and CRK29 peaked at 60 min, and then quickly declined upon flg22 treatment (Additional file 2: Fig. S1G).
CPL3 exerts a profound role in regulating alternative splicing dynamics upon flg22 elicitationCPL3 negatively regulates plant immunity by modulating RNAPII CTD Ser2 dephosphorylation in response to flg22 treatment [20]. Given that RNAPII CTD Ser2 phosphorylation is essential in recruiting splicing components [45], we investigated the role of CPL3 in flg22-regulated AS with the cpl3-3 mutant compared to WT plants for in-depth 3D RNA-seq analysis (Fig. 1A). The cpl3-3 (SALK_094720) has a T-DNA insertion in the 6th exon of AT2G33540 (CPL3), resulting in a truncation of its protein sequence within the FCP homology domain [20] (Additional file 2: Fig. S2A). The mutant was confirmed by PCR genotyping analysis (Additional file 2: Fig. S2B). The growth phenotype of the mutant, observed from 2 to 4 weeks of age, showed no significant differences compared to WT Col-0 plants (Additional file 2: Fig. S2C). The RNA-seq samples of mock- and flg22-treated WT and cpl3-3 seedlings were examined, revealing no significant batch-to-batch variation using the PCA test (Additional file 2: Fig. S1A). Notably, only 1.1% (441 out of 40,932) genes were basal DEGs, and 0.1% (40 out of 40,932) were basal DAS genes between WT and cpl3-3 without flg22 treatment (Additional file 1: Tables S6 and S7), suggesting that CPL3 does not regulate basal transcriptional and AS alterations, consistent with our previous report [20].
In the cpl3-3 mutant, we identified 4562 flg22-DEGs, comprising 2785 upregulated and 1777 downregulated genes, and 691 flg22-DASs with 526 upregulated and 447 downregulated flg22-DTU transcripts (Additional file 1: Tables S2, S3, and S4). Out of the 4562 flg22-DEGs in cpl3-3, 3906 genes (85.6%) overlapped with flg22-DEGs in WT. Similarly, 91.7% of flg22-DEGs in WT overlapped with those in cpl3-3, suggesting that CPL3 only regulates a small portion of flg22-DEGs. Importantly, only about 50% of flg22-DASs overlapped between WT and cpl3-3, which leaves another 50% of flg22-DASs specific to WT or cpl3-3 (Fig. 2A). Similarly, about 50% of IS flg22-DASs were shared between WT and cpl3-3 (Fig. 2B). This analysis indicates that CPL3 plays a more profound role in flg22-regulated AS than its regulation in gene expression.
Fig. 2
CPL3 profoundly affects flg22-triggered alternative splicing events. A CPL3 plays a more important role in flg22-triggered DASs than flg22-triggered DEGs. Venn diagram shows the percentage and gene numbers between up-/downregulated flg22-DEGs (top) and flg22-DASs (bottom) in WT and cpl3-3. The flg22-DEGs were identified based on (|FC|) ≥ 2 and FDR < 0.01. The flg22-DASs were selected based on |ΔPS|≥ 0.1 and FDR < 0.01. B CPL3 regulates isoform-switched DASs. The isoform-switched DASs were identified when a pair of transcripts reversed their relative abundance between mock and flg22 treatments. A Venn comparison plot illustrates the overlap of isoform-switched DASs between WT and cpl3-3 in response to flg22 treatment. C–E Correlation analyses of flg22-DEGs, flg22-DTUs, and flg22-DASs between WT and cpl3-3. The gene expression changes or proportional isoform usages from CPL3-dependent flg22-DEGs (C), flg22-DTUs (D), and flg22-DASs (E) were represented as green, orange, and blue dots with trend lines, respectively. The gene expression changes or proportional isoform usages from CPL3-independent flg22-DEGs, -DTUs, and -DASs were represented as black dots with trend lines. The log2(FC) values were used for flg22-DEGs and flg22-DASs, and the ΔPS values were used for flg22-DTUs. The X-axis and Y-axis values are from WT or cpl3-3, respectively. The trend line equation and the Pearson correlation coefficiencies (R2) between WT and cpl3-3 were labeled. The correlations were analyzed by the Chow test between CPL3-dependent and -independent flg22-DEGs, flg22-DTUs, and flg22-DASs. Non-statistically (ns) and statistically significant differences with p values were indicated in the figure. F GO analysis using 687 of CPL3-dependent flg22-DASs. The statistically enriched gene ontology terms were identified based on the frequency of CPL3-dependent flg22-DASs annotated to their frequency in the genome with the cut-off of fold enrichment ≥ 1 and false discovery rate (FDR) < 0.05. G The total number of flg22-triggered AS events in CPL3-dependent (CPL3-dep) and CPL3-independent (CPL3-indep) flg22-DTUs. The different types of AS events, namely alternative 3′ splicing (A3S), alternative 5′ splicing (A5S), intron retention (IR), and exon skipping (ES), were depicted as red, orange, green, and blue bars, respectively. H The distribution of flg22-triggered AS events in CPL3-dependent and CPL3-independent flg22-DTUs. The number of flg22-triggered AS events across 5′-leader, CDS, and 3′-tailer is indicated by red and blue lines for WT and cpl3-3, respectively. The relative position was calculated as the average of the alternative coordinates of the AS event, scaled by the length of the CDS, and then converted to a percentage based on their full lengths, dividing into 10% windows. The position of the ATG start codon and stop codon are indicated by black dashed lines, separating the 5′-leader and 3′-tailer from the CDS. I Relative isoform abundances of three representative CPL3-dependent flg22-DASs in WT and cpl3-3. The isoform usage (IU) was calculated by the percentage abundance of a transcript compared to the total transcripts of the gene. The β and α represent constitutive and alternative splicing isoforms, respectively. The absolute ΔPS values (in parentheses) and FDR values between mock (white bar) and flg22 treatment (black bar) are indicated at the top of each comparison. Non-statistically significant comparisons (ns) were indicated as gray. The expression levels of individual transcripts were retrieved from RNA-seq data
Heatmap analysis of flg22-DEGs in WT and cpl3-3 classified flg22-DEGs into six groups: UP_WT (genes induced in WT, but not in cpl3); UP_common (genes induced in both WT and cpl3); UP_cpl3 (genes induced in cpl3, but not in WT); DN_WT (genes repressed in WT, but not in cpl3); DN_common (genes repressed in both WT and cpl3); DN_cpl3 (genes repressed in cpl3, but not in WT) (Additional file 2: Fig. S3A). Among the total of 4916 flg22-DEGs in WT and/or cpl3-3, 1010 flg22-DEGs specific to WT or cpl3-3 were designated as CPL3-dependent flg22-DEGs (Additional file 1: Table S8; Additional file 2: Fig. S3B), while the remaining as CPL3-independent flg22-DEGs. The Pearson correlation of expression changes between WT and cpl3-3 for CPL3-dependent flg22-DEGs was lower than that for CPL3-independent flg22-DEGs (R2 values of 0.76 and 0.98, respectively) (Fig. 2C). Moreover, a slope of gene expression change trend-line for CPL3-dependent flg22-DEGs was significantly reduced towards cpl3-3 compared to CPL3-independent flg22-DEGs (p value from the Chow test was 3.3E − 05; Fig. 2C). These results suggest that CPL3 suppresses the expression of upregulated CPL3-dependent flg22-DEGs while enhancing the expression of downregulated CPL3-dependent flg22-DEGs, consistent with our previous results [20]. Apparently, CPL3 has a more profound role in flg22-DEGs than basal DEGs.
Heatmap analysis of 1497 flg22-DTUs in WT and/or cpl3-3 classified flg22-DTUs into six groups: UP_WT (induced IU in WT, but not in cpl3); UP_common (induced IU in both WT and cpl3); UP_cpl3 (induced IU in cpl3, but not in WT); DN_WT (repressed IU in WT, but not in cpl3); DN_common (repressed IU in both WT and cpl3); DN_cpl3 (repressed IU in cpl3, but not in WT) (Additional file 2: Fig. S3C). Among them, 1041 flg22-DTUs specific to WT or cpl3-3 were designated as CPL3-dependent flg22-DTUs, while the remaining as CPL3-independent flg22-DTUs (Additional file 1: Table S9; Additional file 2: Fig. S3D). Similar to flg22-DEGs, the difference in IU of CPL3-dependent flg22-DTUs exhibited a lower correlation than that of CPL3-independent flg22-DTUs (R2 values of 0.75 and 0.95, respectively) (Fig. 2D). Consistently, the slope of the IU difference (ΔPS) trend-line for CPL3-dependent flg22-DTUs showed a significant reduction towards cpl3-3 (p value from the Chow test was 2.2E − 16; Fig. 2D), supporting that CPL3 suppresses IU of upregulated CPL3-dependent flg22-DTUs while enhancing downregulated CPL3-dependent flg22-DTUs. The log2(FC) values of CPL3-dependent flg22-DASs were compared to CPL3-independent flg22-DASs to determine whether the IU changes of CPL3-dependent flg22-DASs are due to their gene expression changes. The Pearson correlation coefficients of CPL3-dependent and -independent flg22-DASs from WT and cpl3-3 were similar (0.97 vs. 0.99; Additional file 1: Table S10; Fig. 2E), suggesting that CPL3 regulates flg22-triggered AS by modulating specific IU rather than controlling gene expression. GO enrichment analysis using 687 of CPL3-dependent flg22-DASs indicates that immune-related terms, such as defense response to other organisms, immune system process, and positive regulation of immune system pathway, were significantly enriched compared to the distribution of GO terms across all genes in the genome (Fig. 2F; Additional file 1: Table S5). Taken together, these results indicate that CPL3 exerts a pronounced effect on flg22-triggered AS events largely independent of its regulation on gene expression level.
Among different types of AS events, alternative 3′ splicing (A3S), alternative 5′ splicing (A5S), intron retention (IR), and exon skipping (ES) were identified as the four major types in flg22-triggered DTUs (Fig. 2G), consistent with a previous report [46]. The total numbers of all four events were substantially increased in CPL3-dependent DTUs compared to CPL3-independent DTUs (Fig. 2G). We further plotted the relative positions of these AS events in flg22-triggered DTUs on a scaled gene structure, divided into 10% windows, consisting of the 5′-leader, coding sequence (CDS), and 3′-tailer. CPL3 has an effect on all four AS events. Apparently, it has a more pronounced role from the end of the CDS to the beginning of the 3′-tailer (Fig. 2H), consistent with its function in dephosphorylating RNAPII CTD Ser2, which is enriched during transcriptional elongation to termination [22,23,24]. Additionally, a quantitative comparison of isoform usage changes between WT and cpl3-3 indicates that among the four AS events, IR showed significantly higher ΔPS values in cpl3-3 than in WT at the CDS and 3′-tailer (Additional file 2: Fig. S3E). These findings suggest that while CPL3 regulates all four major flg22-triggered AS events, it plays a particularly significant role in IR, both in terms of the number of AS events and the ΔPS values at the CDS and 3′-tailer.
We also analyzed in detail a few CPL3-dependent flg22-DASs with previously known functions in plant immunity for their gene structures and protein domains from corresponding DTU transcripts. Interestingly, several membrane-resident proteins related to PTI exhibited CPL3-dependent IU changes upon flg22 treatment. Those include genes encoding RLKs, such as SERK4, CRK29, FRK1, BAK1-INTERACTING RECEPTOR-LIKE KINASE 1 (BIR1), and BACK TO LIFE 2 (BTL2), and a gene encoding calcium transporter AUTO-INHIBITED CA2+ATPASE 12 (ACA12). CPL3 either regulated the IU of both α and β forms of transcripts (SERK4, BTL2, CRK29, FRK1, and ACA12), or only α form of transcripts (BIR1) (Fig. 2I; Additional file 2: Fig. S4A–C). Two MAP KINASES, MPK3 and MPK12, were also among CPL3-dependent flg22-DASs (Additional file 2: Fig. S4A–C). Interestingly, unlike most RLK genes, the constitutive β forms of MPK3 and MPK12 transcripts were induced, whereas their α forms were suppressed upon flg22 treatment (Additional file 2: Fig. S4A).
Since CPL3 plays a crucial role in regulating AS events, we investigated whether CPL3 colocalized with spliceosome components, specifically ARGININE/SERINE-RICH ZINC KNUCKLE-CONTAINING PROTEIN 33 (RSZ33) and SMALL NUCLEAR RIBONUCLEOPROTEIN U1 SUBUNIT 70 (U1-70 k) within the nucleus [47]. RSZ33 and U1-70 K have been widely used as representative markers for spliceosome components, with distinct roles in the mRNA splicing process and their relevance to AS [48, 49]. CPL3-GFP was co-expressed with either RSZ33-RFP or U1-70 k-RFP in Arabidopsis protoplasts and then treated with or without flg22 to examine their colocalization. Notably, the two spliceosome components, RSZ33 and U1-70 k, displayed speckles that did not overlap with the diffused CPL3 signals in the nucleoplasm with and without flg22 treatment (Additional file 2: Fig. S3F). This observation suggests that CPL3 may not directly interact with the spliceosome in regulating AS, but rather operates through the regulation of CTD Ser2 phosphorylation. Consistently, the C-terminal region of CPL3 (CPL3C), which contains the catalytic FCP homology domain [20], dephosphorylated the flg22-triggered CTD Ser2 phosphorylation (Additional file 2: Fig. S3G).
CPL3 regulates the transcription and alternative splicing of DGK5.
The PA biosynthesis gene DGK5, recently implicated in plant immunity [12,13,14], also exhibited CPL3-dependent AS events. While DGK5 displayed a significant upregulation upon flg22 treatment in both WT and cpl3-3, the flg22-induced DGK5 expression was notably higher in cpl3-3 than in WT, suggesting that CPL3 exerts a negative regulatory role on DGK5 expression (Fig. 3A).
Fig. 3
CPL3 negatively regulates flg22-induced DGK5 gene expression and alternative splicing. A Flg22-induced DGK5 expression is elevated in cpl3-3. The gene expression levels of DGK5 were quantified with transcript per million (TPM) values from RNA-seq data (left) and independently confirmed by RT-qPCR (right). Two-week-old seedlings were treated with dH2O or 100 nM flg22 for 60 min for RT-qPCR analysis. Gene expressions of DGK5 were normalized to UBQ10, and data are shown with mean ± S.D. from three biological repeats (n = 3). Data were analyzed by unpaired two-tailed Student’s t-test between WT and cpl3-3. The p values are indicated at the top of the figure. B Diagrams of gene structures and protein domain annotations for three DGK5 isoforms. The read coverage for the DGK5 gene from each sample was visualized using an Integrative Genomics Viewer (IGV) (top). Introns are represented by solid lines, while exons and alternatively spliced regions are depicted by black and purple boxes, respectively (middle). Two pairs of primers distinguishing DGK5β and DGK5α isoforms from the last exon are indicated by blue and red arrows, respectively. While the primers amplifying DGK5β could also amplify DGK5γ, the expression levels of DGK5γ were negligible with less than 1 TPM in the RNA-seq analysis. The intron retention (IR) site is indicated by the dashed lines on the top of the panel. The alternative 3′ splice site (A3S) is indicated by the dashed lines on the bottom of the panel. The constitutive protein denoted as DGK5β (blue) contains the DGK catalytic domain (DGKc), DGK accessory domain (DGKa), and calmodulin-binding domain (CBD) (bottom). DGK5α (orange) lacks the CBD, and DGK5γ only contains partial DGKc. C The flg22-induced upregulation of DGK5β and DGK5α is elevated in cpl3-3. Transcript levels of DGK5β and DGK5α were quantified with TPM values from RNA-seq data. The mock and flg22 treatments are depicted as opened and closed bars, respectively. The fold changes were analyzed by comparing TPM values between mock and flg22 treatments and indicated at the top of each comparison in parentheses. Data were analyzed by unpaired two-tailed Student’s t-test between WT and cpl3-3, and the p values are indicated at the top of each comparison. D The IUs of DGK5β and DGK5α in WT and cpl3-3 upon flg22 treatment. The IU was calculated by the percentage abundance of each transcript compared to the total transcripts of the gene. The absolute ΔPS values (in parentheses) and FDR values between mock (opened bar) and flg22 treatment (closed bar) are indicated at the top of each comparison. Data were analyzed by unpaired two-tailed Student’s t-test between WT and cpl3-3, and the p values or non-statistically significant differences (ns) are indicated at the top of each comparison. E Flg22-triggered transient induction of DGK5β and DGK5α in WT and cpl3-3. Two-week-old seedlings were treated with 100 nM flg22 for the indicated times for RT-qPCR analysis. Transcript expressions of DGK5β and DGK5α were normalized to UBQ10, and data are shown with mean ± S.D. from three biological repeats (n = 3). Data were analyzed by unpaired two-tailed Student’s t-test between WT and cpl3-3 mutant. The p values between WT and cpl3-3 are indicated at the top of each comparison. Non-statistically differences comparisons (ns) are indicated as gray. F Multiple elicitors induce the transcript levels of DGK5β and DGK5α. Two-week-old seedlings were treated with mock, 100 nM flg22, elf18, or scoop12 for 60 min for RT-qPCR analysis. Data are shown as mean ± S.D. from four biological repeats (n = 4) analyzed by one-way ANOVA followed by Tukey’s test with the p values indicating statistical difference. G The α-DGK5 antibody detects endogenous DGK5β and DGK5α proteins. Total proteins were extracted from 2-week-old WT and dgk5-1 seedlings, followed by immunoblotting using an α-DGK5 antibody with Rubisco (RBS) stained by Coomassie brilliant blue (CBB) as a loading control. The polyclonal α-DGK5 antibody was generated by using full-length DGK5 proteins as an antigen from rabbits
Interestingly, forty-two transcript isoforms of DGK5 were identified from AtRTD3 and were categorized into three protein groups based on their translation and functional domain annotation by FGENESH (www.softberry.com). The full-length protein sequence, referred to as DGK5β, contains DGK catalytic domain (DGKc), DGK accessory domain (DGKa), and calmodulin-binding domain (CBD) [12, 13, 50]. The truncated isoform lacking the CBD was named DGK5α, similar to other DGK homologs in plants [50, 51], and another truncated isoform containing only partial DGKc was DGK5γ (Additional file 2: Fig. S5A). The DGK5α isoform arises from the intron retention at the last intron, resulting in CBD truncation at the carboxyl terminus (Fig. 3B). The DGK5γ isoform underwent alternative splicing at the 3′ end of the first intron, leading to the alternative 3′ splice site (A3S) and resulting in a truncated protein at the DGKc domain (Fig. 3B). The aggregated TPM values from RNA-seq data were utilized to examine the total transcript levels for each isoform. The expression levels of DGK5γ were negligible, with less than 1 TPM in both mock and flg22 treatment conditions (Additional file 2: Fig. S5B). Conversely, expression levels of DGK5β and DGK5α were significantly induced by flg22 treatment (Fig. 3C). Moreover, the flg22-induced expression of DGK5β and DGK5α was further enhanced in cpl3-3 compared to WT, suggesting that CPL3 negatively regulates the expression of both DGK5β and DGK5α (Fig. 3C). Additionally, we assessed the IUs of two DGK5 isoforms by examining the proportion of their transcripts relative to the total DGK5 transcripts (Additional file 2: Fig. S5C). While the IUs of DGK5β and DGK5α were not significantly altered in WT upon flg22 treatment, their IUs were significantly and oppositely changed in cpl3-3, further supporting the regulatory role of CPL3 in DGK5 AS events (Fig. 3D).
RT-qPCR analysis with primers specific to each isoform (Fig. 3B) confirmed the flg22-triggered induction of both DGK5β and DGK5α. This induction was transient, peaking at half an hour post-treatment (hpt) and gradually returning to basal levels by 3 hpt (Fig. 3E). Notably, CPL3 regulated DGK5α levels across all time points following flg22 treatment (Fig. 3E). Additionally, the expression level of DGK5α was about 10 times higher than DGK5β, both with and without flg22 treatment. In addition to flg22, the MAMP elf18 and the phytocytokine scoop12 also significantly induced the expression of both DGK5β and DGK5α (Fig.
Comments (0)