Cell cultureiPSCs
The usage of human induced pluripotent stem cells (iPSCs) was approved by the Human Gamete, Embryo and Stem Cell Research (GESCR) Committee at UCSF.
The NGN2-inducible piggyBac-containing iPS3 cell line (hDFn 83/22 iNgn2#9 [iPS3]) was originally described in Nehme et al. The NGN2-inducible piggyBac-containing iPSC line KOLF2.1 J was obtained from Jackson Laboratory. Stem cells were cultured in StemFlex Medium (Life Technologies, A3349401) on Geltrex-coated (LDEV-Free, hESC-Qualified Geltrex basement membrane matrix, Gibco, A1413301) tissue cell culture plates at 37ºC in 5% CO2. ROCK inhibitor (Y-27632, custom synthesis) was added to media at the time of passaging and removed the next day.
NGN2 neuron differentiation
Patterned induced neurons were differentiated through a combination of doxycycline to rapidly induce NGN2 expression and small molecule patterning factors as previously described (Pintacuda et al.) [32], with the exceptions of excluding normocin or geneticin selection. On day 5–7 post-induction, neurons were passaged via accutase treatment onto plates coated with PLO/laminin (poly-L-ornithine hydrobromide, Sigma, P3655; laminin, Life technologies, 23,017,015) in neuron maintenance medium [neurobasal media (Gibco), 0.2% N-2 supplement (Life technologies), 2% B-27 supplement (Life technologies), 1% Glutamax, 1% MEM non-essential amino acids (NEAA) solution (Gibco)] supplemented with 1:10,000 doxycycline, neurotrophic factors: 1:10,000 brain-derived neurotrophic factor (BDNF), 1:10,000 ciliary neurotrophic factor (CNTF), 1:10,000 glial cell-derived neurotrophic factor (GDNF) (Bio-techne) and, to remove any residual proliferating cells, EdU (5-ethynyl-2’-deoxyuridine) or FUdR (5-Fluoro-2′-deoxyuridine) at 2 uM was briefly added to cultures. On day 7 and onwards, neurons were fed by replacing half of the media with neuron maintenance medium supplemented with neurotrophic factors, every 2–3 days.
Cell lines
HEK 293 T cells were cultured in DMEM with 10% FBS (Takara, 631,106 or Fisher Cytiva HyClone, SH3008803). For the transfection of HEK cells, plasmids expressing DPRs fused with GFP or GFP alone, as previously described (Lee et al.) [14], were diluted in Opti-MEM media and transfected with polyethylenimine (PEI, Sigma, 408,727) in 10-cm plates for proteomic experiments or Lipofectamine 3000 reagent (Invitrogen) according to the manufacturer’s standard protocol for additional experiments. Cells were washed in PBS and then briefly trypsinized and immediately pelleted and flash frozen for storage at -80C for subsequent proteomics, or directly lysed, three days post transfection.
All cell lines were routinely tested for mycoplasma with the MycoAlert PLUS Mycoplasma Detection Kit (Lonza, LT07-703), and were negative.
Phospho-proteomics
The phospho-proteomics and bulk proteomics was performed at Harvard Center for Mass Spectrometry (HCMS). S-trap mini columns were used (Protifi, NY) according to manufacturer protocol. Briefly, samples were resolubilized in 5% SDS for reduction and alkylation, further digested in buffer trypsin (Promega) for 5 h. The digested samples were enriched by High-Select™ TiO2 Phosphopeptide Enrichment Kit (Thermo-Fisher) according to the vendor’s instructions. Enriched phosphopeptides were labeled with TMT16plexPRO (Thermo-Fisher) according to manufacturer protocol. Sample fraction was submitted for single LC–MS/MS experiment that was performed on a Q Exactive HF (Thermo, MA) equipped with 3000 Ultima Dual nanoHPLC pump (Thermo). The Q Exactive HF was operated in data-dependent mode for the mass spectrometry methods. The mass spectrometry survey scan was performed in the Q Exactive HF Orbitrap in the range of 450 –950 m/z at a resolution of 6 × 104, followed by the selection of the twenty most intense ions TOP10 ions were subjected to HCD MS2 event in Orbitrap. The fragmentation isolation width was 0.8 m/z, AGC was 50,000, the maximum ion time was 150 ms, normalized collision energy was 32 V and an activation time of 1 ms for each HCD MS2 scan was set. For phospho-peptides with multiple PTMs, phospho-residues may not necessarily be unambiguously assigned.
Bulk proteomics
Following the trypsin digest, samples were labeled with TMT6plex (Thermo-Scientific, MA) according to manufacturer protocol. Then, all labeled samples been combined into single sample, and then further desalted and separated by HipH cartridges (Thermo-Scientific, MA) into 20 fractions. Fractions were dried in speedvac (Eppendorf, Germany) and resolubilized on 0.1% formic acid in water buffer for injections into mass spectrometer for analysis of all fractions, which were submitted for single LC–MS/MS experiment as described above for phospho-proteomics.
Mass spectrometry analysis
Raw data were analyzed in Proteome Discoverer 3.0 (Thermo Scientific) software with Byonic 3.5 node and ptmRS node. Assignment of MS/MS spectra was performed using the Sequest HT algorithm by searching the data against a protein sequence database including all entries from the Human Uniprot database. A MS2 spectra assignment false discovery rate (FDR) of 1% on protein level was achieved by applying the target-decoy database search. Filtering was performed using a Percolator (64bit version) (Kall et al., 2008). For quantification, a 0.02 m/z window centered on the theoretical m/z value of each the six reporter ions and the intensity of the signal closest to the theoretical m/z value was recorded. Reporter ion intensities were exported in result file of Proteome Discoverer 2.4 search engine as an excel tables. The exact place of phosphor moiety was analyzed by ptmRS program (Taus et al., 2011). Differentially expressed proteins between sample groups were analyzed using R script programs based on Bioconductor (https://www.bioconductor.org/). Statistical analysis for differentially expressed proteins was based on peptide level to find changes of phosphor moiety presence in peptides that are statistically significant between two sets. For bulk proteomics, Proteome Discoverer searches were performed with set modification of TMT labeling to each N-terminus of the peptide and side chain of lysine as well as carbamidomethyl modification to cysteine amino acids, and variable modification to allow methionine oxidation and protein N-terminus acetylation. All searches were filtered to 0.1% FDR at protein and peptide levels. Gene ontology enrichment analysis was performed on the g:Profiler toolset server.
Modulation of gene expression
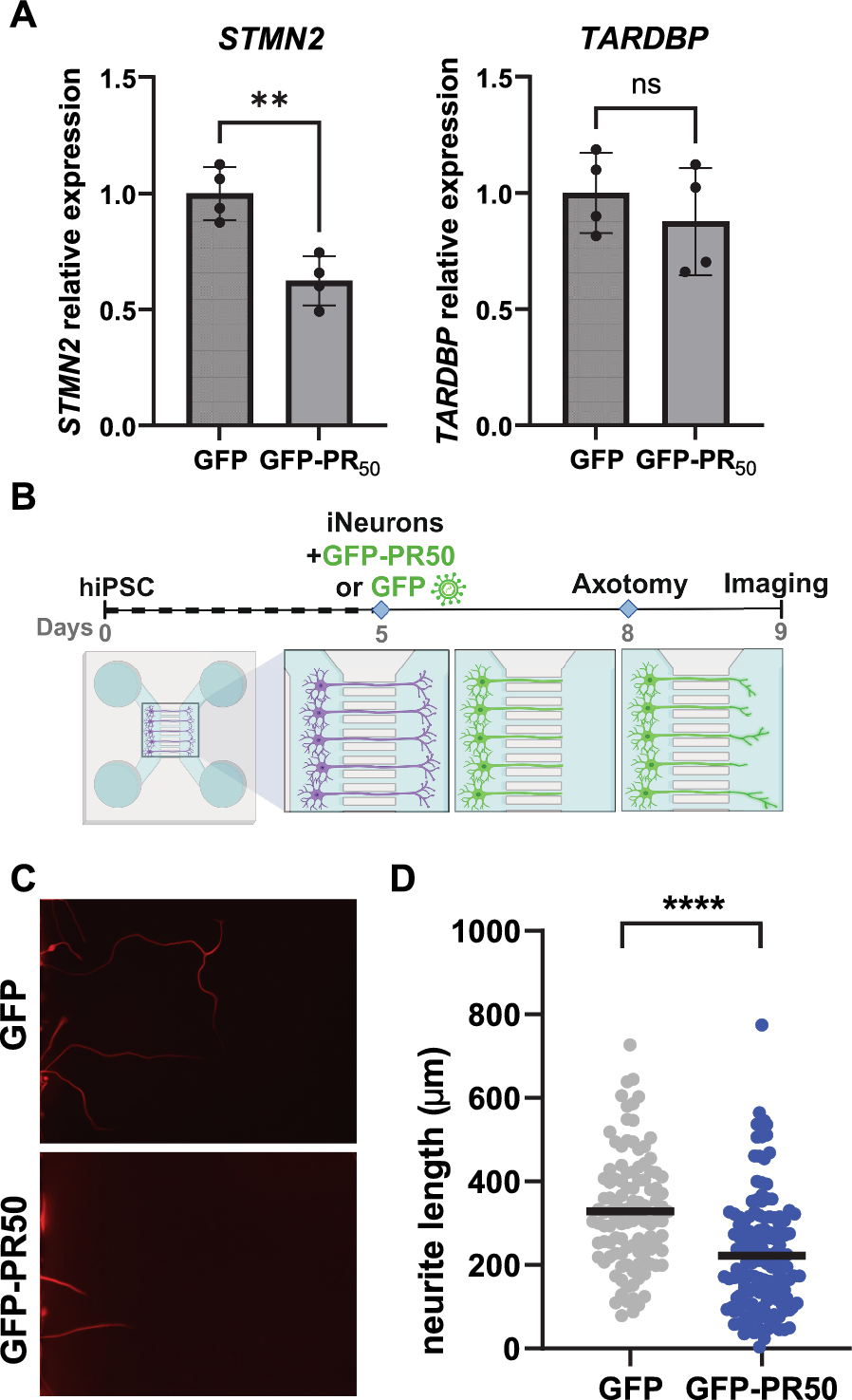
Pooled siRNAs (SMARTpool) targeting transcripts encoding RNA binding proteins, STMN2, and non-targeting control siRNAs (ON-TARGETplus) and individual siRNAs (ON-TARGETplus modified to minimize off-target effects) were obtained from Horizon Discovery-Dharmacon. Lyophilized siRNAs were resuspended in diluted 5X siRNA Buffer (Horizon Discovery-Dharmacon, B-002000-UB-100) at 20 uM, aliquoted, and stored at -80C. For knockdown experiments, induced neurons were transfected using Lipofectamine RNAiMAX (Invitrogen) with pooled siRNAs (SMARTpool) as a single dose and then harvested for RNA 72 h later, or with individual siRNAs in two doses (0, 48 h) and then harvested at 96 h later. The most effective individual siRNA targeting SRSF7 was compared to a non-targeting control siRNA for functional studies. Total RNA was prepared from cells or neurons using the RNeasy Plus Mini Kit (Qiagen, 74,134) or RNeasy Plus Micro Kit (Qiagen, 74,034), respectively, according to the manufacturer’s protocol.
Immunoblotting
For Western blotting, cells and neurons were lysed in Pierce IP lysis buffer (Thermo Scientific) supplemented with a phosphatase and protease inhibitor cocktail (Roche PhosSTOP and cOmplete, Mini, EDTA-free protease inhibitor cocktail). Cleared lysates were combined with sample buffer containing DTT and run on gradient proteins gels (Mini-PROTEAN TGX Precast Gels, Biorad) and transferred using the iBlot2 system. Following blocking, primary antibodies were applied- anti-STMN2 (rabbit, Proteintech, 10,586–1-AP), anti-SRSF7 (Rabbit, Proteintech, 11,044–1-AP), anti-GAPDH (clone 6C5 mouse, Millipore Sigma, MILL-CB1001), anti-neuron-specific beta-III Tubulin (Tuj-1; mouse, R&D systems, MAB1195), anti-TDP-43 C-terminal (rabbit, Proteintech, 12,892–1-AP)- then anti-mouse or anti-rabbit LICOR IRdye secondary antibodies. Signal detection and quantification was performed on the LICOR ODYSSEY system.
Quantitative PCR
Reverse transcription of RNA to complementary DNA was performed using the iScript™ Advanced cDNA Synthesis Kit (Bio-Rad, 1,725,038). Quantitative RT-PCR (qRT-PCR) was performed with diluted cDNA, primers (synthesized by Integrated DNA Technologies), and the Maxima SYBR green qPCR master mix 2X (Thermo Fisher, K0253) with reactions run on a Real-Time PCR System (CFX96, Bio-Rad). All reactions were run in technical triplicates on the thermocycler and then averaged. Levels of all genes assayed were normalized to GAPDH expression unless noted otherwise. Normalized gene expression is presented as relative to the control sample. Primer pair sequences were obtained from prior publications or the MGH PrimerBank, and sequences are provided in the supplementary table.
Lentiviral poly-PR and synthetic poly-PR
Lentiviral constructs to express STMN2-GFP, GFP, GFP-PR50 were described in Linares et al [33]. SRSF7-GFP (pLenti) to produce lentivirus was obtained from OriGene. The expected nuclear localization of this epitope-tagged SRSF7 was confirmed via fluorescent microscopy. For the transduction of neurons, high-titer lentiviral preparations from 293 T cells were performed by Alstem. Due to the higher expression of GFP alone, GFP-PR50 virus was used at twice the volume of GFP. PR20 was synthesized by Peptide 2.0 with purity > 95%, confirmed by HPLC, and dissolved in DMSO. Vehicle (DMSO) or PR20 peptide at a non-toxic concentration was added directly to neuron cell culture media.
Axotomy assay
NGN2 hiPSC-derived neurons at day 5 post-differentiation were cultured on microfluidic devices (XONA Microfluidics SND150), mounted on glass coverslips pre-coated with 0.1 mg/mL poly-L-ornithine in 50 mM Borate buffer and 5ug/mL laminin, at a density of 100,000 neurons per device. GFP-PR50 or GFP lentivirus transduction was carried out in suspension during cell replating. A complete media change was executed the following day. Axotomy was conducted three days after plating in the microfluidic chambers by repeated vacuum aspiration following perfusion of 1X PBS in the axonal chamber. After axotomy, SiR-tubulin dye (NC0958386, Cytoskeleton) was diluted in fresh neuronal media. This dye allows for the observation of neurite morphology without the need for cell fixation, thereby avoiding potential confounding effects on cell morphology. Subsequently, images were taken 24 h post-axotomy using a widefield microscope at 20X magnification. The quantification of regrowing neurites, based on sirTubulin staining, was conducted using NeuronJ on ImageJ.






Comments (0)