
Remember me

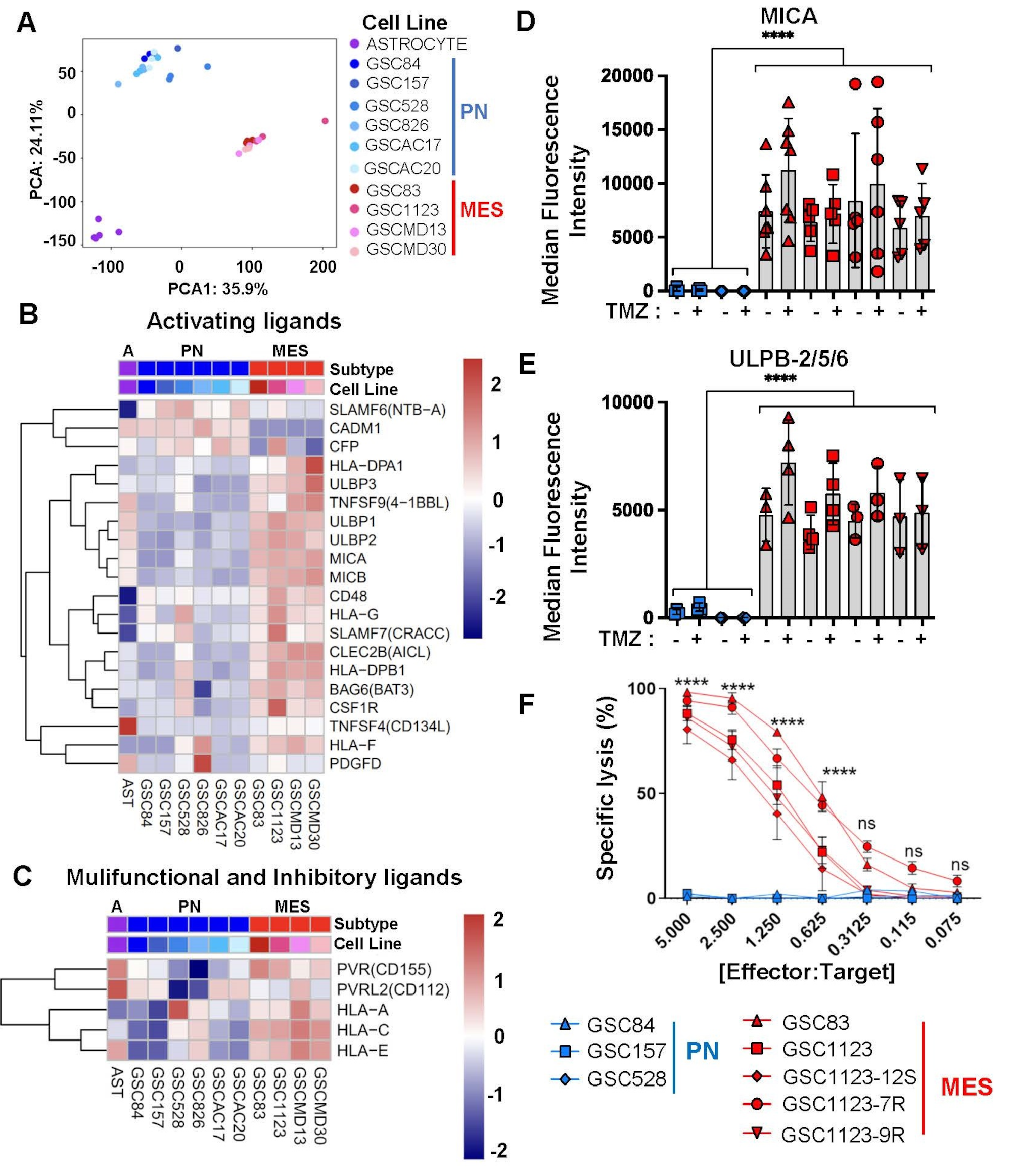




Given the aggressive and recurrent nature of HGG we sought to understand whether the susceptibility of tumour-driving GSCs to NK-mediated killing may explain the apparent failure of innate immune surveillance in this disease. For this purpose, we interrogated the available gene expression databases of PN- or MES-GSCs isolated from brain tumours comprising multiple HGG subtypes [30], along with control normal human astrocytes [30], for known activating, multifunctional, or inhibitory ligands able to engage critical NK cell receptors [23, 41]. Notably, global transcriptomes revealed a clear separation of these cell lines into distinct clusters around either astrocytes, PN-GSCs, or MES-GSCs, thus highlighting their molecular heterogeneity (Fig. 1A). While there was some variability in the expression of mRNA for specific NK activating ligands between individual GSC lines, the levels of these transcripts were almost uniformly higher for MES-GSCs than for their PN-GSC counterparts, regardless of the nature of the original HGG isolate. This difference included MICA, MICB, ULBP1 and ULBP2, which are the canonical activators of the NKG2D receptor on NK cells (Fig. 1B) [23]. A similar, albeit less obvious, trend was observed in the case of multifunctional and inhibitory NK ligands (Fig. 1C and fig. S1). It is noteworthy that the molecular classification of hitherto characterized GSCs into PN and MES populations is predicated on their transcriptomic similarity to corresponding subtypes of grade IV HGG with wild type status of the IDH1/2 genes, currently classified as glioblastoma (GBM) [30, 51]. It should also be noted that GBM also includes a classical (CL) molecular subtype, in addition to PN and MES tumours [51] but GSCs in our series did not contain a corresponding transcriptome [20, 30]. To assess whether these bulk GBM transcriptomes would also differ with respect to NK ligands (as do GSCs) we performed in silico analysis of their corresponding mRNA profiles using the Cancer Genome Atlas (TCGA) database (fig. S2A). Indeed, MES GBM samples expressed considerably more of some (MICA and MICB), but not all transcripts encoding NK ligands relative to their PN counterparts, and there were also differences in MICA between CL and PN GBM tissues (fig. S2-S3). These observations suggest that MES transcriptome is enriched in NK ligands, both in bulk GBM and in isolated GSCs.
Fig. 1
Differential expression of NK cell ligands and susceptibility to NK cell-mediated cytotoxicity between patient-derived mesenchymal and proneural glioma stem cell populations. (A) Principal component analysis (PCA) of total transcriptomes of indicated cells: proneural GSCs (PN), mesenchymal GSCs (MES) and astrocytes. (B-C) In silico analysis of mRNA expression levels (microarrays) for NK cell ligands in a panel of PN and MES GSCs and Astrocytes. Activating NK ligand transcripts (B) and transcripts for multifunctional NK ligands (C). (D-E) Differential cell surface expression of NK ligands interacting with cytotoxic NKG2D by MES-GSCs and PN-GSCs (FACS); MICA (D) and ULBP-2/5/6 (E). GSCs were assayed with or without pretreatment with temozolomide (TMZ). MICA and ULBP-2/5/6 protein levels were expressed as median fluorescent intensity +/- SEM, for n = 3–5 independent repetitions, p values were determined for group comparisons by unpaired two-tailed t-test, p < 0.0001 (F) NK cell specific target cell (GSC) lysis analyzed by bioluminescence assay. GSCs were incubated with NK92MI human immortalized NK cells for 4 h at indicated ratios. Experiments were performed at least 3 times, **** p < 0.0001 as determined for group comparison between PN and MES time points by one way ANOVA with Tukey’s multiple comparison test +/- SEM
We have also examined PN- or MES-GSCs for NK ligand protein levels and functionality (Fig. 1D-F). We carried out some of these experiments in the presence or absence of temozolomide to account for the realities of GBM therapy (fig. S4). In this regard, flow cytometry analysis of NKG2D ligands, such as MICA and ULBP2/5/6 (Fig. 1D, E; fig. S5A-C and fig. S6), and DNAM1 ligand, PVR/CD155 (fig. S5C, S7), demonstrated their preferential and abundant expression by MES-GSCs [29], but not by PN-GSCs. MICB was not detectable on the surface of either GSC subtype (fig. S5B) [10]. While TMZ exposure was suggested to increase the responsiveness of GBM cells to NK-mediated cytotoxicity [10, 50] we observed no major changes in levels of key NK cell ligands (Fig. 1D, E; fig. S5A, B) in the presence of physiological and biologically effective concentrations [21, 35] of the drug (fig. S4). Also under in vivo growth conditions, in GSC xenografts, the levels of MICA remained markedly elevated in MES-GSC tumours relative to PN-GSC lesions, and did not observably change following the exposure of tumour bearing mice to TMZ, as determined by quantitative flow cytometry (Fig. 2; fig. S8).
Fig. 2
NK cell ligand expression by intracranial GSC xenografts in the presence or absence of temozolomide therapy. (A) MICA immunohistochemical staining of proneural (GSC528) and mesenchymal (GSC1123 and GSC83) intracranial xenografts in SCID mice (magnification = 200X (size bar = 50 μm). (B-C) FACS analysis of NK ligand expression in dissociated GSC1123 intracranial tumours. MICA (B) and ULBP-2/5/6 (C) expression by cancer cells in mice either untreated (Control) or 48 h post temozolomide (TMZ) treatment. Mean fluorescence intensity +/- SEM (right side panels in B and C) and corresponding FACS histograms; Unstained GSC123 cells (blue), MICA- or ULBP-2/5/6-stained GSC1123 control cells from culture (red), and three independent stained GSC1123 cell populations (n = 3) isolated from untreated (light green, dark green and pink) or TMZ treated tumours (light purple, dark purple and dark blue). Statistical analysis by unpaired T-test, ** p < 0.01
Finally, we used the bioluminescence cytotoxicity assays [25] to explore more directly the effect of the engineered human NK cell line (NK92MI) against a panel of the aforementioned GSCs in culture. Remarkably, while all MES-GSC lines were efficiently killed by NK92MI cells across a wide range of effector-to-target ratios, their PN-GSC counterparts remained completely unaffected by human NK effectors (Fig. 1F, fig. S9, tab. S1). Overall, these results suggest that the intrinsic molecular subtype of GSCs represents a notable determinant of their cellular sensitivity to NK-mediated cytotoxicity, with MES-GSCs being especially susceptible to such killing.
Elevated sensitivity of mesenchymal glioma stem cells to NK-mediated surveillance post chemotherapy in vivoIn order to determine whether differential sensitivity to NK cell-mediated killing in vitro translates into corresponding antitumour responses in vivo, PN-GSCs and MES-GSCs were used to generate subcutaneous xenografts in strains of mice, which were either proficient (SCID) [5, 16, 47] or deficient (NSG) for NK cell activity (Fig. 3; fig. S10) [42, 55]. Both GSC subtypes efficiently triggered tumour formation in either strain of mice, with PN-GSCs exhibiting an expectedly slower growth kinetics and aggressiveness [20, 30]. We have earlier documented that in these settings, the exposure of tumour bearing NK-deficient NSG mice to TMZ produced a profound, but transient disease remission followed by a relapse of TMZ resistant and incurable lesions [20]. In order to examine the possible role of NK cells in this disease evolution process, NSG and SCID mice were inoculated with either MES-GSCs (GSC1123, GSC83) or PN-GSCs (GSC528) and upon tumour establishment the animals were exposed to a single dose of TMZ (120 mg/kg i.p.). We observed that, as expected, in NK-deficient NSG mice this therapy resulted in a profound but transient tumour regression followed by a relapse of both MES-GSC and PN-GSC-driven lesions after approximately 40–50 days. This pattern was also recapitulated in NK-proficient SCID mice inoculated with PN-GSC cells (Fig. 3A-B; fig. S10B-E). However, in the case of SCID mice harbouring MES-GSC tumours the post-TMZ tumour relapse was completely aborted and the mice survived beyond 200 days without any evidence of active disease (Fig. 3A; fig. S10B). In contrast, PN-GSC-driven tumours continued to relapse in SCID mice (Fig. 3B; fig. S10D). To further ascertain that these effects can be attributed to the suggested capacity of mouse NK cells to recognize human cancer cell targets [7] and kill them [54] we isolated NKp46/NCR1-positive lymphocytes from mouse splenocyte preparations [53] (fig. S11) and performed bioluminescent cytotoxicity assays using human mesenchymal GSC1123bfpLuc glioma cells as targets. As expected, mouse NK cells were robustly cytotoxic against these GSCs (Fig. 3C). In several cases of surviving SCID mice a faint bioluminescent signal of residual MES-GSC cells was observed several months post TMZ therapy in the absence of progressive disease (fig. S12A-C), suggesting a possible contribution of tumour cell dormancy to anticancer effects of endogenous NK cells [15]. Collectively, these observations are consistent with the notion that MES-GSCs (but not PN-GSCs) are sensitive to NK cell effects in vivo. Moreover, since both NSG and SCID mice support the unperturbed tumour growth, TMZ-dependent tumour depopulation appears to be required for endogenous murine NK cells to exert an effective surveillance over the residual MES-GSCs, resulting in GSC subtype-specific obliteration of disease re-initiation.
Fig. 3
Impact of endogenous mouse NK cells on post-chemotherapy recurrence of mesenchymal glioma stem cell xenografts. (A) Endogenous NK cell proficiency of SCID mice supports eradication of mesenchymal glioma stem cell (GSC1123) - driven subcutaneous xenografts by single dose of temozolomide (TMZ; 120 mg/kg); data expressed as mean +/- SD; (B) Endogenous NK cell deficiency of NSG mice is associated with post-TMZ relapse of GSC1123 subcutaneous xenografts (mean +/- SD); (C) Purified mouse NK cells efficiently kill GSC1123 mesenchymal glioma stem cells in vitro in a time dependent manner (BLI assay with readings conducted at 5 h and 22 h post NK treatment; mean value +/- SEM); (D) Experimental design to explore the effects of pharmacological NK inhibition (Asialo-GM1) on progression of mesenchymal GSC subcutaneous xenografts post TMZ-mediated tumour depopulation; (E) Asialo-GM1 treatment aborts TMZ-mediated GSC1123 tumour eradication in NK-proficient SCID mice– bioluminescent images; (F) Inhibition of endogenous NK cell cytotoxicity (Asialo-GM1) counteracts pro-survival effects of TMZ in NK-proficient SCID mice. Error is represented by SEM
Pharmacological targeting of NK cells overrides tumour eradication post chemotherapyIn order to further explore the role of innate immunity in curative effects of TMZ in NK-proficient SCID mice the animals bearing MES-GSC xenografts were depleted for NK cells using anti-AsialoGM1 (anti-NK) antibody [32] (Fig. 3D-F). Two weeks prior to cancer cell inoculation, SCID mice were randomized and subjected to weekly i.v. injections of either anti-Asialo-GM1, or vehicle for a total of 9 weeks. On day 14 of this cycle, all mice were subcutaneously inoculated with cancer cells (GSC1123) and once the tumours became established (500 mm3) a single dose of TMZ (120 mg/kg) was injected i.p. followed by monitoring tumour responses (BLI, palpation) (Fig. 3D). In keeping with the aforementioned observations, TMZ exhibited a curative effect in control NK cell-proficient SCID mice, as indicated by disease free survival that extended beyond 150 days for the entire cohort. Notably, anti-Asialo-GM1 treatment reversed this effect almost completely with the majority (7/8) of mice exhibiting gradual tumour recurrence resulting in endpoint within less than 56 days post TMZ (Fig. 3E-F; fig. S13). These results further suggest that in SCID mice harbouring MES-GSC xenografts the endogenous NK activity is functionally required for tumour eradication and long-term survival post TMZ-dependent cancer cell depopulation. Thus, in mice depleted for NK cells either genetically (NSG strain) or pharmacologically (Asialo-GM1) the post-TMZ long-term survival is disrupted by tumour recurrence.
Recruitment of NK cells into the tumour mass in the course of temozolomide-induced regressionIn order to visualise cellular events associated with TMZ-induced tumour regression, followed by either relapse, or cure in NSG or SCID mice, respectively, MES-GSC xenografts were subjected to serial histological examination (fig. S14A, B). As expected, a single dose of TMZ triggered a progressive loss of cellularity and dramatic shrinkage of the tumour mass in both NSG and SCID mice over a period of 2–3 weeks, after which cancer cell deposits became virtually undetectable until recurrence (in NSG mice only, fig. S14B). During this time tumour remnants in SCID mice exhibited a biphasic influx of mouse NCR1-positive NK cells peaking on days 3 and 14 post TMZ (fig. S15), with infiltrating NK cells forming thick rims around necrotic tumour regions and at the boundaries of regressing cancer cell deposits and surrounding stromal tissue. While scarce NCR1 staining was also detected in tumours harboured by NK-deficient NSG mice, no robust infiltration has occurred in this setting in agreement with the underlying NK cell maturation defect [1]. However, in both strains of mice the microenvironment of regressing tumours attracted other bone marrow derived, mouse, CD45-expressing cells (fig. S16). Microvascular density highlighted by CD31 staining was not visibly different between tumours in SCID and NSG mice and even increased somewhat, as expected, with ongoing depopulation of viable cancer cells between capillaries (fig. S17). Overall, these observations suggest that in both NK cell-proficient and -deficient mice the MES-GSC xenografts underwent protracted and nearly complete regression post TMZ treatment. This process was paralleled by waves of NK cell infiltration, which were largely restricted to SCID mice.
Intracranial microenvironment restricts NK cell impact on post-chemotherapy survival of tumour bearing miceThe brain microenvironment uniquely restricts the access of drugs [37] and immune cells [49] to the tumour site resulting in their diminished anticancer activities. To explore this factor in the context of NK-sensitive MES-GSCs xenografts (Fig. 4A, B), these cells were inoculated intracranially [20] into NK cell-proficient SCID mice. Upon tumour establishment (as defined by the BLI signal) the mice received TMZ intraperitoneally at either a standard experimental dose of 120 mg/kg, or at increased dose of 200 mg/kg (Fig. 4B), established to compensate for reduced drug penetration into the brain [21, 35, 37]. Strikingly, neither of these TMZ treatment regimens recapitulated the dramatic prolongation in survival of tumour bearing SCID mice observed when the tumours were grown subcutaneously (Fig. 4A). Even at the higher dose of TMZ (200 mg/kg) only approximately 50% of mice survived longer than 150 days, while the remaining mice experienced tumour relapse before day 100 (Fig. 4B). These results are in agreement with reports suggesting a poor presence of NK cells in the brain tumour parenchyma [49], which is also consistent with our observations suggesting a limited representation of mouse CD45+, or NCR1 + cells in MES-GSC intracranial xenografts in SCID mice prior to treatment (fig. S18). Thus, the curative potential of NK cells during post-TMZ evolution of MES-GSC tumours is likely curtailed by poor access of these cells into the brain.
Fig. 4
Intracranial delivery of exogenous NK cells blocks post-temozolomide recurrence of mesenchymal glioma xenografts. (A) Kaplan-Meier curves indicating responsiveness of subcutaneous GSC1123 xenografts to TMZ treatment (120 mg/kg, i.p) in SCID mice. (B) Kaplan-Meier curves documenting relative refractoriness of intracranial GSC1123 xenografts to TMZ therapy (120 or 200 mg/kg i.p.) in SCID mice. (C) Experimental design of intracranial NK92MI therapy. (D) BLI representative scans of SCID mice in indicated treatment groups: untreated, NK92MI, TMZ + Media or TMZ + NK92MI. (E) Kaplan-Meier curves of GSC1123 tumour bearing mice for corresponding treatment groups show efficacy of TMZ and NK92MI combination. Statistical analysis was conducted by Curve comparison using the Gehan-Breslow-Wilcoxon test. Untreated group (n = 13 mice, 2 repeats), NK92MI treated group (n = 5 mice, 1 repeat), TMZ + media group (n = 15 mice, 4 repeats), TMZ + NK92MI group (n = 6 mice, 2 repeats). (F) Distribution of exogenous NK92MI cells in the brain of mice with GSC1123 xenografts: untreated (left) and TMZ + NK92MI treated mice (right) showing infiltration of NK92MI cells (brown) into the choroid plexus (CP) of lateral ventricle (LV) and meninges (M) 150 days post treatment
Intracranial NK cell therapy following temozolomide-mediated cytoreduction eradicates orthotopic mesenchymal glioma stem cell xenograftsAlthough the mechanism of endogenous NK cell exclusion from the brain tumour parenchyma remains poorly understood, one avenue to circumvent this barrier is through a direct intracranial immunotherapy [6, 24]. We set out to explore whether this approach could re-open the post-TMZ window of curative opportunity in the case of MES-GSC-driven tumours. MES-GSCs were inoculated intracranially into SCID mice and tumours were allowed to emerge (as BLI signal), at which point the mice were exposed to TMZ (120 mg/kg) followed two days later by injection of live human NK-like cell line, NK-92MI (Fig. 4C). These cells are engineered to remain in an activated state by constitutive expression of interleukin 2 (IL2) [46] and were approved by Food and Drug Administration (FDA) for experimental anticancer therapy in clinical trial settings [33]. Moreover, in our hands NK92MI cells exhibited a robust in vitro cytotoxicity against MES-GSC lines, but not against their PN-GSC counterparts (Fig. 1F). SCID mice were used as tumour recipients, as in these mice intracranial MES-GSC xenografts are not efficiently infiltrated by endogenous NK cells and are not eradicated following TMZ chemotherapy alone (Fig. 4B). Remarkably, the intracranial injection of NK92MI cells following TMZ chemotherapy radically changed the natural history of MES-GSC (GSC1123) tumours, resulting in tumour regression and long-term survival (> 150 days). This outcome is in contrast to the lethality of mice that received no therapy, or were injected only with NK92MI cells, or with TMZ alone, with “media” to serve as a surgical control (Fig. 4D, E; fig. S19). Histological examination of brains from all experimental groups showed infiltration of some mouse CD45 + cells and a scarce influx of endogenous NCR1 + mouse NK cells into the regressing tumour masses regardless of treatment (fig. S18). A global morphological overview documented end stage tumours in all groups except for the TMZ + NK92MI treated mice in which no tumours were detectable (fig. S19). Moreover, intracranial mesenchymal xenografts (GSC1123, GSC83) continued to express high levels of NK ligands, such as MICA and ULBP-2/5/6 in situ (Fig. 2; fig. S8A), in contrast to their proneural counterparts (GSC528), which remained negative for these molecules. This expression was somewhat increased by, but did not depend upon, the exposure to TMZ (fig. S8B, C). Overall, these results suggest that neither endogenous NK cells, nor exogenous NK92MI cells alone, even when injected intracranially, are able to prolong survival of MES-GSC brain tumour bearing SCID mice, unless NK92MI therapy is combined with TMZ-mediated tumour depopulation.
Curative effects of lethally irradiated NK92MI cells combined with TMZ in mice harbouring mesenchymal glioma stem cell xenograftsAlthough NK92MI cells are FDA approved for human trials and exerted curative effects in MES-GSC-driven brain tumours, they are immortalised, transformed and thereby may pose inherent biosafety risks. Notably, NK92MI cells were detectable in brains of ostensibly cured (brain tumour-free) mice earlier inoculated with MES-GSCs (GSC1123; Fig. 4F and fig. S20). In some experiments these cells exhibited signs of neoplastic expansion throughout cerebral structures (fig. S21). One approach to decouple cytotoxicity of NK92MI cells from their proliferative potential relies on sublethal irradiation [28]. Exposure of NK92MI cells to radiation doses ranging from 2 Gy to 10 Gy was undertaken aiming to establish conditions that would maintain a sustained cell viability combined with cell cycle arrest (fig. S22A, B), which was achieved at 5 Gy. Indeed, following irradiation with 5 Gy NK92MI cells retained their structural integrity and viability for up to 7 days in vitro (fig. S22C), and they also remained selectively cytotoxic to MES-GSCs (fig. S22D). In order to test whether such irradiated NK92MI cells (NK92MI-IRs) are still able to prevent recurrence of TMZ treated MES-GSC xenografts, the respective cell lines (GSC1123, GSC83) were inoculated intracranially into SCID mice. Established tumours were then exposed to systemic TMZ (120 mg/kg), followed by intracranial NK92MI-IR therapy (Fig. 5A). Strikingly, this protocol resulted in aborted recurrence and long term survival of mice bearing GSC83 (5/5)(Fig. 5E and fig. S23 and S24B) or GSC1123 (4/6) (Fig. 5, B-D) xenografts, while neither TMZ alone (+ media) nor NK92MI-IRs alone were able to achieve similar effects. There were no remarkable differences in the infiltration of the tumour mass with endogenous mouse CD45-positive bone marrow-derived cells, or NCR1-positive mouse NK cells, as a function of NK92MI cell injection (fig. S25A, B). GSC xenografts contained some mouse NCR1-positive cells in low abundance in all groups, with mice treated with TMZ alone being an intriguing, but presently unexplained exception (fig. S25C). Give
Comments (0)