Collection of tissue samples
This study included 30 female KOA patients who underwent knee arthroplasty in the Department of Orthopaedic Surgery of the First Hospital of Jilin University from August 2020 to June 2021 and from September 2024 to December 2024. The inclusion criteria were as follows: (1) the diagnosis of KOA met the American College of Rheumatology’s diagnostic criteria for OA; (2) no drug use or drug test in the past month; (3) denied any history of knee trauma and surgery; (4) female patient. The exclusion criteria were as follows: (1) did not voluntarily sign the informed consent; (2) secondary KOA; (3) received intra-articular injection therapy within the past 6 months; (4) had a history of knee trauma or knee joint infection; (5) autoimmune diseases or diseases that affect other parts of the body, such as asthma, systemic lupus erythematosus, rheumatoid arthritis, and tumors. Those who met al.l 4 inclusion criteria and did not meet any one of the exclusion criteria were included in the study, and those who met any one of the 5 exclusion criteria were not included in the study. Female patients with KOA were divided into MetS-KOA and nMetS-KOA according to the NCEP-ATPIII criteria for the diagnosis of MetS in Asians [16], which gave 15 cases each of MetS-KOA and nMetS-KOA. The patients signed the informed consent form and we obtained consent for the study from the Medical Ethics Committee of the First Hospital of Jilin University (No. 2015 − 284). The patient tissue samples were then collected during the operation. Characteristics of the study population are shown in Supplementary Table S1.
After fasting for at least 8 h before surgery, 8 ml blood was collected. The samples were sent to the laboratory within 10 min, and centrifuged at 3000 rpm for 10 min at 4 °C. The serum was dispensed and numbered, and then stored in the − 80 °C freezer of the tissue specimen bank. During the total knee arthroplasty, the resected synovium of suprapatellar capsular was collected. The synovium used for pathological experiments was immediately fixed in a specimen bottle containing 10% neutral formalin. The synovium used for cell extraction was put into a sterile 15 ml tube, 10 ml of FBS-free DMEM high-glucose medium was added, and samples were quickly transferred to the laboratory within 20 min for cell extraction. The synovium was sent to the laboratory at 4 °C and then packed and numbered into the − 80 ℃ refrigerator of the tissue specimen bank for future use.
Isolation and culture of FLS
Isolation of FLS was done by enzymatic digestion [16]. The isolated synovium was washed three times with sterile PBS, and the synovium was cut into tissue fragments of approximately 2–3 mm3 in size. After washing, the synovium fragments were collected by centrifugation at 1500 rpm for 5 min, followed by addition of 2.5 mg/ml collagenase type I for digestion on a constant temperature shaker at 37 ℃ for 2 h. The digested solution was filtered, then centrifuged at 1500 rpm for 5 min. And the pellet was washed three times with PBS. The cell pellet was resuspended in high-glucose DMEM containing 10% FBS and 1% penicillin-streptomycin solution, transferred to a culture flask, and cultured in a 5% CO2 cell incubator at 37 ℃. The FLS used in this experiment were the 3rd to 6th generation.
Pathology experiments
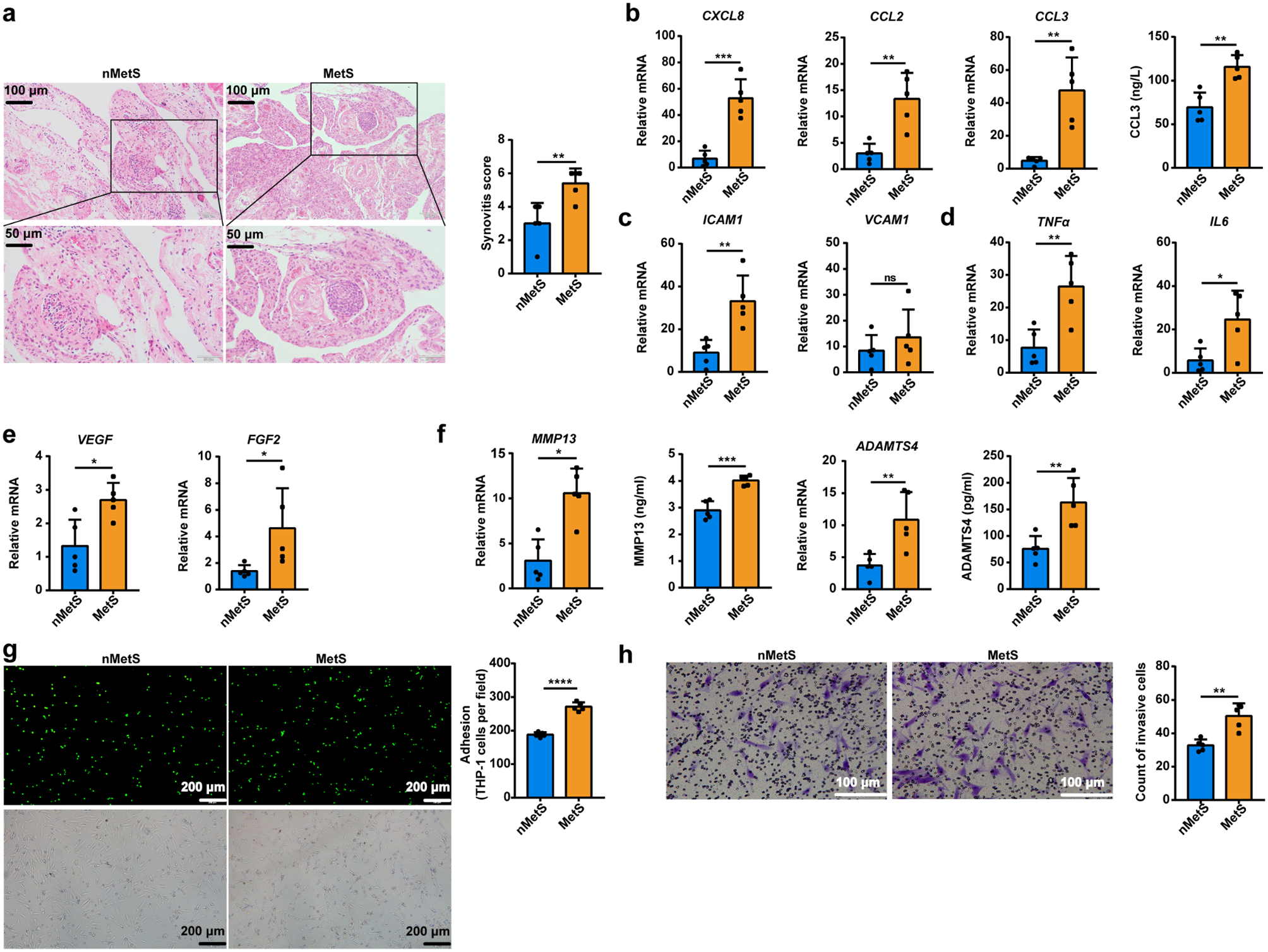
The collected synovium was fixed in 10% neutral formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E) and the relevant antibodies. Images were acquired under an OLYMPUS upright microscope using an Olympus D71 image acquisition system. At least 3 images were randomly taken for each tissue section, and statistical analysis was performed on the average level. The degree of synovitis was assessed according to the histopathological scoring criteria for synovitis [16]. The semi-quantitative assessment (ranging from 0 (absent) to 3 (intense)) was used based on three features of synovitis (proliferation of the synovial lining layer, cellular density of the synovial matrix, and leukocyte infiltration), each scored separately. The total score of synovitis was 0–1, with no synovitis; 2–4 points, low-grade synovitis; 5 to 9 points, high degree of synovitis. Antibodies used for immunohistochemistry were as follows: CAP1 (Abcam, ab155079), dilution ratio 1:200; ready-to-use HRP-polymer anti-rabbit IgG (MXB Biotechnologies, KIT-5920). ImageJ was used to measure the mean optical density.
Cell counting kit CCK-8
MetS-KOA-FLS were seeded on a 96-well plate at 5000 cells/well. The cells were cultured for 24 h to completely adhere, then various concentrations of resistin (0 ng/ml, 100 ng/ml, 200 ng/ml, 500 ng/ml) were added to the culture plate, with three replicate wells per condition. The 96-well plate was put back into the incubator, and 10 µl of CCK-8 solution was added to the wells at 6 h, 12 h, 24 h, 48 h, and 72 h, followed by further incubation for 1–4 h. The absorbance at 450 nm was measured with a microplate reader, and the cell viability was calculated.
Cell stimulation experiments
In the serum stimulation experiment, 1 ml of serum from 5 MetS patients were put into the same 15 ml centrifuge tube and mixed. nMetS-KOA-FLS were incubated for 24 h with DMEM containing 1% penicillin-streptomycin solution which was mixed with either the medium containing 5% MetS serum (serum stimulation group), or 5% FBS (control group).
In the pathway intervention experiment, 10 nM TAK-242 (MedChemExpress, HY-11109), 2.5 µM H-89 (Beyotime, S1643-1 mg), or 2.5 µM KG-501 (MedChemExpress, HY-103299) were pre-incubated with MetS-KOA-FLS for 1 h and then co-incubated with 200 ng/ml resistin for 24 h. For 2-Deoxy-D-glucose (2-DG) and Etomoxir (Eto) treatments, 10 mM 2-DG (Solarbio, D8930) or 100 µM Eto (Sigma, SIG-236020-5MG) were pre-incubated with MetS-KOA-FLS for 2 h and then 200 ng/ml resistin was added for a further 24 h.
Transfection
FLS were resuspended in high-glucose DMEM without penicillin-streptomycin solution, seeded in 12-well plates at 5 × 104 cells/well, and incubated for 24 h before transfection. The siRNA stock was diluted in high-glucose DMEM without penicillin-streptomycin solution to a final concentration of 50 nM for transfection. Lipofectamine 2000 (Invitrogen, 11668-019) was diluted in high-glucose DMEM without penicillin-streptomycin solution at a ratio of 1:50, and left to stand for 5 min at 25 ℃. The lipofectamine 2000 and siRNA diluents were mixed at a 1:1 ratio (100 µl each), gently pipetted 3–5 times, and left at 25 ℃ for 20 min. The medium in the 12-well plate containing cells was changed in advance, and 800 µl of high-glucose DMEM without penicillin-streptomycin solution was added to each well with 200 µl/well of the above transfection mixture. After 4–6 h of culture, the medium was replaced with fresh complete medium. Forty-eight hours after transfection, cells were treated with resistin and/or harvested for qPCR or WB to detect the silencing efficiency of siRNA at the gene or protein level. CAP1-siRNA was purchased from GENERAL BIOL in China, and the detailed sequence information is shown in Supplementary Table S2.
Western blotting and antibodies
The cells were washed twice with 4 °C pre-cooled PBS, then lysis on ice for 40 min. After ensuring complete cell lysis, the lysate was centrifuged at 12,000 rpm for 20 min at 4 °C and the supernatant was collected. Protein sample concentration was determined using a BCA kit (KeyGEN BioTECH, KGP902-500T). Loading buffer was added and mixed well with the sample, followed by centrifugation, and incubation in a water bath at 100 °C for 10 min. Approximately 20 µg per protein sample was added to 10% SDS-PAGE gel for electrophoresis to separate proteins. The resulting protein bands were transferred to nitrocellulose filter membrane. The blots were then incubated with primary antibody overnight at 4 ℃. Finally, fluorescent secondary antibody was added and incubated for 1 h, then developed and photographed in a Li-COR fluorescence imager. The gray value of the band was measured by AlphaEaseFC software, and statistical analysis was performed. Antibodies used were as follows: HK2 (Cell Signaling Technology, #2867), LDHA (Cell Signaling Technology, #3582), CPT1A (Cell Signaling Technology, #12252), CAP1 (Abcam, ab155079), TLR4 (WanleiBio, WL00196), CREB (Cell Signaling Technology, #9197), pCREBser133 (Cell Signaling Technology, #9198), β-actin (Cell Signaling Technology, #4970), Dylight 549, goat anti-rabbit IgG (Abbkine, ABB-A23320). The dilution ratio of primary antibody was 1:1000, and the dilution ratio of fluorescent secondary antibody was 1:10000.
Quantitative real-time polymerase chain reaction (qRT-PCR)
RNA was prepared using AG RNAex Pro RNA extraction reagent (ACCURATE BIOLOGY, AG21102). Reverse transcription PCR of RNA was performed using 5X All-In-One RT MasterMix cDNA Synthesis Kit (Applied Biological Material, G490). qPCR was performed on a Biorad CFX96 machine with the SYBR green-based program using the SYBR® Green Premix Pro Taq HS qPCR Kit (ACCURATE BIOLOGY, AG11701). Normalization was performed using the 2 −ΔΔCt method. PCR primers were designed using the online primer tool Primer3Plus. Detailed sequence information of primers is listed in Supplementary Table S3.
Invasion assay
Growth factor reduced (gfr) Matrigel (Becton, Dickinson and Company, 356231-10 ml, 8.9 mg/ml) was diluted 1:1 with DMEM. An 8.0 μm transwell chamber was coated with 30 µl of the diluted Matrigel and placed at 37 ℃ for 30 min. Next, serum-free cultured nMetS-KOA-FLS and MetS-KOA-FLS (2 × 104 cells per group) were seeded in transwell chambers (the method was the same for seeding cells with or without stimulation with resistin, 2-DG, and Eto). After 72 h of cell culture, the cells that did not pass through the filter membrane of the transwell chamber were wiped from the top surface with a cotton swab. The remaining cells on the bottom surface of the chamber were fixed in 4% (w/v) paraformaldehyde for 20 min, then stained with 0.1% (w/v) crystal violet and counted under the microscope. Invasion values were expressed as the mean number of migrating cells (200×) at the bottom of the chamber per 3 microscopic fields.
Adhesion assay
FLS were seeded in 20 mm glass bottom dishes at 1 × 105 cells/dish. After the cells were completely attached, they were treated according to the experimental plan, and collected by centrifugation at 3 × 105 cells/dish one hour before the end of the experimental treatment. BCECF AM was added to the THP-1 cell suspension to a final concentration of 10 µM. THP-1 cells were incubated in a 37 °C, 5% CO2 sterile cell incubator for 1 h in the dark, and then added to a culture dish at 3 × 105 cells/dish and incubated with FLS for 6 h. The unadherent THP-1 cells were removed by gentle washing with PBS 2–3 times, and images were taken on the OLYMPUS upright microscope using the Olympus D71 image acquisition system. Invasion values were expressed as the average number of adherent cells (100×) per microscope view at the bottom of the dish. Three fields of view were counted per dish. Adherent THP-1 cells were counted using ImageJ and the results were statistically analyzed using Graphpad.
ELISA
Debris or dead cells were removed from the cell culture supernatant by centrifugation, and then CCL3 (Nanjing Jiancheng Bioengineering Institute, H110), MMP13 (Nanjing Jiancheng Bioengineering Institute, H459-1), or ADAMTS4 (CUSABIO, CSB-E11848h) were detected in the cell culture supernatant according to the l instructions of the ELISA kit manufacturers.
Annexin V-FITC/7-AAD
1 × 105 cells per group were seeded in 12-well plates. In addition to the experimental group and the control group, 3 flow detection control groups (including the normal group, the 7-AAD single staining group, and the Annexin V-FITC single staining group) were set up to adjust the fluorescence compensation, remove spectral overlap, and set the position of the cross gate. After treatment, cells were digested with EDTA-free trypsin, and then collected by centrifugation at 1500 rpm for 5 min. The cells were washed by adding pre-cooled PBS buffer, and then centrifuged at 1500 rpm for 5 min to collect the cells, and washed again. The cells were resuspended in 100 µl of 1× Binding Buffer. Annexin V-FITC and 7-AAD (5 µl of each) were added to the experimental group and the control group, and the three flow cytometry control groups were prepared accordingly (normal cell group; no dye, 7-AAD single staining group; 5 µl of 7-AAD, Annexin V-FITC single staining group; 5 µl of Annexin V-FITC). After mixing, the samples were incubated at room temperature for 15 min in the dark. The flow cytometer NovoCyte Flow Cytometer (Agilent Technologies, CA, US) was used for detection, with FITC as the abscissa and 7-AAD as the ordinate to draw a two-color scatter plot.
Statistical analysis
Continuous variables were expressed as mean ± standard deviation (SD), and independent samples t-tests or paired t-tests were used for comparative analysis. The WB bands were measured by AlphaEaseFC software and converted into continuous variables for quantitative analysis, and then the differences between groups were compared. Experiments were independently repeated three times. P < 0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism 7.0 software and Microsoft Excel.






Comments (0)