
Remember me
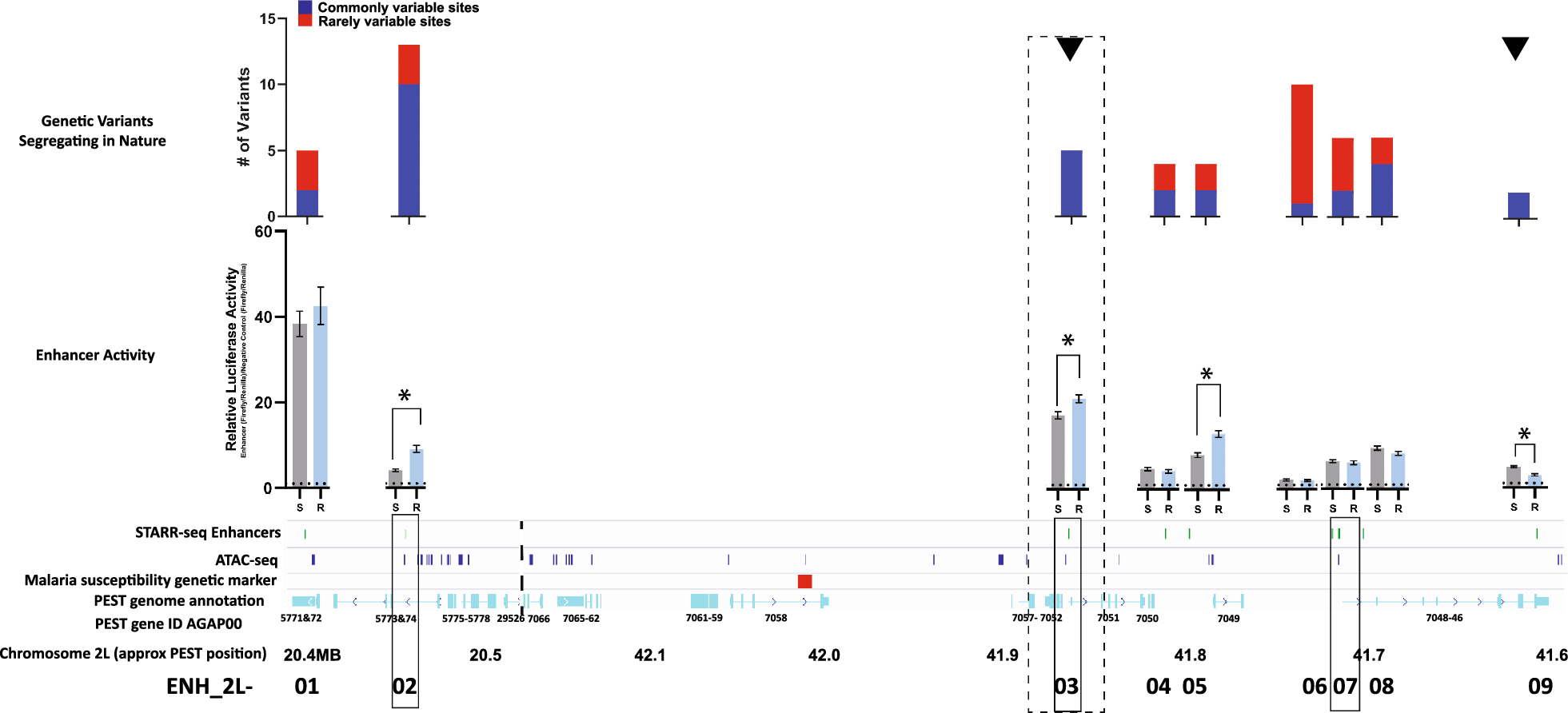
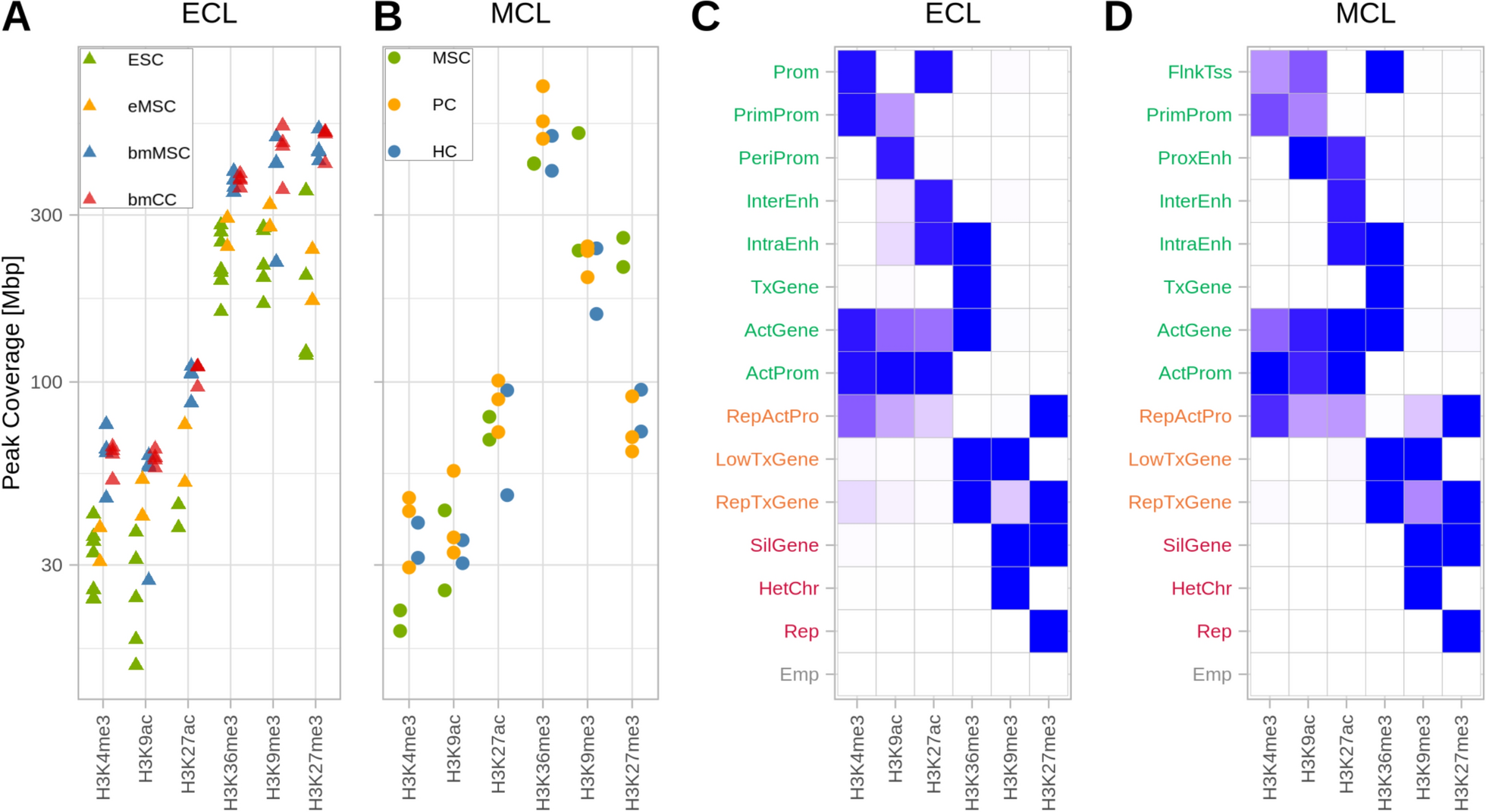
To generate an epigenetic profile of the chondrogenic lineage we analyzed two ChIP-Seq datasets representing (1) the early differentiation stages from ESC to mesenchymal and chondrogenic cells, and (2) later differentiation stages from MSC to proliferating and hypertrophic chondrocytes. The dataset of early differentiation stages (ECL; early chondrogenic lineage) consists of 4 cell lines, which cover a broad differentiation spectrum but do not include specific chondrocyte cell types. Due to the lack of publicly available data of early mouse differentiation stages, in which the same set of histone marks has been investigated as in our previous study [71], we used human datasets of the Roadmap consortium: (1) embryonic stem cells (ESC), (2) MSC differentiated from ESC (eMSC), (3) bone marrow-derived MSC (bmMSC) and (4) chondrogenic cells differentiated from bmMSC (bmCC). To focus on direct differentiation steps within the chondrogenic lineage the second dataset (MCL; mature chondrogenic lineage) represents the differentiation of MSC derived from ribs of 6-week-old mice [69] into the defined chondrocyte subtypes of PC and HC, which were isolated by FACS sorting [71]. The chondrogenic potential of MSC has been confirmed by Wu et al. [69]. For all cell types, we analyzed 6 histone modifications covering the promoter region (H3K4me3, H3K9ac, H3K27ac), the transcribed gene body (H3K36me3), and repressed genomic regions (H3K27me3 and H3K9me3).
As a first step, we evaluated the genomic coverage of the histone marks. Peak calling detected narrow peaks for the promoter marks, while H3K36me3, H3K27me3, and H3K9me3 demarcated broader genomic regions. Accumulating the peak length of each modification revealed a similar pattern in the respective cell types, with lower genomic coverage for promoter marks and considerably higher coverage for the broad marks. In the ECL data, the more differentiated, bone marrow-derived cell types clustered together, showing a generally higher level of modification than the ESC-derived samples (Fig. 1A). The cell types of the MCL showed a similar coverage for most histone modifications except for the coverage of the repressive mark H3K27me3, which was notably higher in MSC than in PC and HC (Fig. 1B).
Fig. 1
The coverage of histone marks and ChromHMM states are similar in both datasets. A-B Genomic coverage of the epigenetic marks for each cell type and replicate. Points are spread out horizontally by small displacements to reduce overplotting. C-D Emission probabilities of the 15-state ChromHMM model, based on the consensus data of the replicates. The colors of the row names categorize the states into four groups: activating (green), repressed-active (orange), repressive (red), and empty (grey)
To estimate the overall similarity of the data we calculated the Jaccard index of each pair of samples (Supp.Fig. B3A-B). In both datasets, the distribution of activating promoter marks was highly similar between each other, within and between the individual cell types. The marks characterized by broad peaks agreed well between cell types but differed from each other and from the promoter marks. Furthermore, for all marks of the ECL dataset, the ESC-derived cell types (ESC, eMSC) showed better agreement with each other than with the bone marrow-derived cells (bmMSC, bmCC), possibly reflecting their different biological origin (Supp.Fig. B3A). In the MCL dataset, the genomic localization of the repressive marks H3K27me3 and H3K9me3 was considerably different in MSC compared to PC and HC (Supp.Fig. B3B).
Understanding the role of epigenetic modifications at a distinct genomic position requires determining the combination of marks at the given location. For each dataset (ECL and MCL), we trained a hidden Markov model (“ChromHMM model”) on the consensus data of each histone mark per cell type (see flowchart in (Supp.Fig. B1A). Models with different numbers of chromatin states were evaluated based on their log-likelihood and the clarity of biological interpretation of the states (Supp.Fig. B4A,D). For each dataset, a 15-state model was chosen, and the respective states were annotated based on the mark emission, the localization at distinct genomic regions (genome, gene, exon, CpG island, TSS, (Supp.Fig. B4B,E)), the distribution around the anchor points (TSS and TES Supp.Fig. B4C,F), and the expected regulatory role of the accumulated marks [21, 24, 33, 62]. The models derived from each dataset were robust, with 13 out of 15 states occurring in both datasets (Fig. 1C-D). Besides non-decorated regions [empty states (Emp)] we defined six common states linked to active gene expression covering the promoter region [active gene (ActGene), active promoters (ActProm), and primed promoters (PrimProm)], the transcribed gene body [intragenic enhancers (IntraEnh), transcribed genes (TxGene)], and enhancer regions [intergenic enhancers (InterEnh)]. In addition, two activating states covering the promoter region differed between the datasets: The peripheral promoter (PeriProm) and promoter (Prom) states in the ECL data were substituted with the proximal enhancer (ProxEnh) and flanking TSS (FlnkTSS) states in the MCL data. In both datasets, three states were composed of only repressive marks [silenced genes (SilGene), heterochromatin (HetChr), and repressed regions (Rep)], and three states were characterized by a combination of activating and repressive marks [low transcribed genes (LowTxGene), repressed transcribed genes (RepTxGene) and repressed active promoter (RepActPro)].
Among these states, the RepActPro state is especially intriguing as it can be regarded as a variant of the classical bivalent state (H3K4me3/H3K27me3), which has been described to mark lineage-specific genes in ESC [4, 8]. Surprisingly, although ESC are part of the ECL data, the bivalent state did not emerge in the described 15-state ChromHMM model. It was, however, detected if only ESC were used to train a separate 15-state model (Supp.Fig. B5A-B). Nevertheless, in this model, the RepActPro state was also detected although with a lower prevalence than in the full ECL model.
While overall the distribution of states was quite similar between the ECL and MCL model, single states show a different emission of individual marks. For example, the low emission of H3K9me3 on the MCL RepActPro state was not included in the respective ECL state. As the ChromHMM models were derived from different species, this might indicate a species-specific effect. To test whether such a difference is critical for the identity of the state, we applied the ECL-derived, human ChromHMM model to the murine MCL replicates and compared it to the murine model by calculating the Jacquard index. The murine FlnkTSS and ProxEnh states weakly colocate with different promoter and enhancer states of the humanized model, likely reflecting the high dynamics in promoter-associated activating states. The 13 identically annotated states including the RepActPro state were almost exclusively assigned to the same genomic region in both models despite the slight difference in states emission (Supp.Fig. B6). Additionally, visualization of exemplary genes carrying the RepActPro state (Supp.Fig. B7) supported the similarity between the human and murine ChromHMM model.
As demonstrated above, the consensus data led to robust chromatin state models. At the same time, the use of consensus data does not account for the variance between replicates. However, this variance is important to assess whether a given difference in state coverage between two cell types simply reflects the background variation, or is linked to a specific role in one cell type. To include the variance between replicates the 15 ChromHMM states were mapped to the individual replicates of each mark and cell type (see flow chart in Supp.Fig. B1A). In the ECL data, the state coverage was clearly dependent on the cellular origin (ESC or bone-marrow-derived) (Supp.Fig. B5A), whereas the MCL data displayed an increased coverage of the H3K27me3-containing SilGene state in MSC (Supp.Fig. B5C). While the observed differences in state coverage were consistent with the pattern of the individual marks, it remains unclear if they reflect a cell type-specific, epigenetic decoration, related to lineage differentiation, or if they are due to technical variances of independently obtained data.
To distinguish between these alternatives, we analyzed the coverage of ChromHMM states on tissue-specific genes relative to the coverage on all protein-coding genes (Supp.Fig. B8). Based on GO terms [3, 15], we defined two groups of tissue-specific genes, a group of 78 mesenchymal and a group of 105 chondrogenic genes. Groups of 40, 50, or 100 random draws of background genes and a published group of 270 housekeeping genes [29] served as controls (Supp.Fig. B8).
Fig. 2
The relative coverage of ChromHMM states reveals systematic differences of epigenetic coverage between cell types. Coverage in base pairs of a given state (row) on a given gene set (column) was plotted against the coverage on all genes. A shows data of the ECL and (B) of the MCL. The black line (slope = [length of all background genes] / [length of all genes within a set], intercept= 0) delineates an equal proportional coverage of the background and the gene set. Deviation from the line indicates enrichment (below the line) or depletion (above the line) of the state in the respective gene set. See Supp. Fig. B8 for all combinations of gene sets and states
As expected, the state coverage on the randomly drawn genes did not deviate markedly from the background, especially for higher numbers of random genes. Similarly, the relative state coverage of the housekeeping and tissue-specific (mesenchymal, chondrogenic) genes did not differ considerably between the distinct cell types of each dataset (Fig. 2). Most notably the SilGene state, which has an increased prevalence in MSC compared to PC and HC, did not deviate from the background if relative state levels were considered. This strongly indicates that the increased coverage was not related to an epigenetic function, but rather represented a generally elevated decoration in MSC or a technical difference in data acquisition.
While the relative prevalence was similar between the cell types, closer inspection revealed deviations from the background for distinct states. This was most obvious for housekeeping genes, which were enriched in activating states (ActGene, ActProm) in all cell types, while repressive states like the SilGene state were underrepresented. Similar enrichment and depletion patterns were seen for mesenchymal and chondrogenic genes (Fig. 2). Interestingly, in the ECL data the relative prevalence of states of active transcription, like the ActGene, IntraEnh, and TxGene state, correlated with cellular maturation, especially for chondrogenic genes, switching from being underrepresented to an above-average decoration in further differentiated cells (Fig. 2, Supp.Fig. B8).
Taken together, our data emphasize the importance of a relative data analysis, in order to distinguish unspecific and potentially artificial effects from more nuanced, ‘true’ effects, associated with gene expression during cell differentiation.
Taking the relativistic approach one step further, we next aimed to understand how specific alterations in the epigenetic profile (transitions) were related to cell differentiation. For a quantitative answer that accounts for sample variation, we built a Bayesian model that calculates transition probabilities. In short, the model determines the probability with which a transition between a pair of states occurs between two cell types on a gene in the background or a gene of a given set. The direction of transition between replicates of the same cell type (e.g. MSC to MSC) was assigned arbitrarily and reflect the dynamics of decoration within the cell type. The direction between cell types was based on the direction of differentiation: from ESC to eMSC to bmMSC to bmCC for the ECL data, and from MSC to PC to HC for the MCL data. These transitions reflect changes that are linked to differentiation. The model passed all checks for convergence and validity (see Methods, Supp.Fig. B9, Supp.Fig. B10, and Supp.Fig. B11). The transition probabilities were expressed in terms of the common-language effect size value (CLES), a score between 0 and 1. Values below and above 0.5 indicate depletion and enrichment of transitions in the gene set in comparison to the background, respectively (Fig. 3A, Supp.Fig. B1B for graphical explanation).
Fig. 3
Systematic enrichment of activating marks during lineage establishment. Individual effect sizes for selected transitions of housekeeping, chondrogenic, and mesenchymal genes (CLES color key at the bottom left). Left and bottom axes represent the origin and target states, respectively, with state groups empty (gray), repressive (red), and activating (green). A Exemplary transition of the ECL data on housekeeping genes from the TxGene state to the IntraEnh state. This active-to-active state transition was enriched between all analyzed cell types. The enrichment was particularly strong from ESC into more mature cells and from every cell type into bmCC. B Selection of transition with drastic shifts of the epigenetic pattern, from empty or repressive states to activating states. See Supp.Fig. B12 for all combinations of states, cell types, and gene sets
We first analyzed the control gene sets of 40, 50, and 100 randomly drawn genes. In these control sets, most transitions had effects with CLES values between 0.45 to 0.55. Especially with higher gene numbers barely any effects or discernible patterns were visible. In contrast, much stronger effects with clear trends towards enrichment and depletion were observed for the housekeeping and the tissue-specific gene sets (Supp.Fig. B12). As expected for housekeeping genes, the most notable enrichments with CLES values higher than 0.6 were predominantly found on transitions between activating states covering the promoter region and the gene body (Fig. 3A, Fig. 4), while strong depletions were mainly limited to transitions involving repressive and empty states, emphasizing the continuous expression of housekeeping genes throughout differentiation. Interestingly, with the maturation of ESC-derived to bone marrow-derived cell types of the ECL, we observed an enrichment of transitions from empty to transcribed states (ActGene and TxGene). In contrast, transitions into partially activated promoter states (PrimProm, PeriProm, InterEnh) were depleted (Fig. 3B). Such enrichments were not observed in the more specialized cell types of the MCL data (Supp.Fig. B12), indicating that the decoration of housekeeping genes with activating marks is strengthened during early differentiation stages and maintained thereafter.
In contrast to housekeeping genes, drastic state transitions, like empty to activating or repressive to activating were more prevalent in the tissue-specific gene sets, especially in the ECL, which covers more distant developmental stages (Supp.Fig. B12). For mesenchymal genes, the transitions into activating states were primarily enriched with the differentiation into eMSC and bmMSC, while the epigenetic activation of chondrogenic genes becomes more pronounced with the differentiation into chondrogenic cells (bmCC) (Fig. 3B). In contrast, in the MCL data transitions from repressed and empty into activating states were rarely detected while transitions between various activating states were more defined, especially on chondrogenic genes (Supp.Fig. B12).
One striking difference between housekeeping and tissue-specific mesenchymal and chondrogenic genes was the behavior of the RepActPro state, which carries a combination of several activating marks and the repressive H3K27me3. While the persistence of this state (self-transition of RepActPro to RepActPro) was depleted in the continuously expressed housekeeping genes, it was slightly enriched on mesenchymal and strongly enriched on chondrogenic genes (Fig. 4). Furthermore, in the tissue-specific genes, we also detected transitions from the RepActPro state into activating states (ActGene and ActProm) and vice versa, indicating a regulatory function of such transitions.
Fig. 4
The formation and breakdown of the RepActPro state are directly linked to cellular differentiation. Housekeeping genes reveal a consistent pattern in ECL and MCL data, with clear enrichments between the activating promoter states (ActGene and ActProm; top left corners in the individual sections) and depletions for the persistence of the RepActPro state (bottom right corners in the individual sections). For chondrogenic and mesenchymal genes there is an enrichment in the formation of the activating states by losing H3K27me3 in the ECL (RepActPro to ActGene/ActProm; bottom rows in the individual sections). In contrast, in the MCL the formation of the RepActPro state by gaining H3K27me3 is more pronounced (ActGene/ActProm to RepActPro; right column in the individual sections). See Supp.Fig. B12 for all combinations of states, cell types, and gene sets
Specifically, in the ECL dataset, we found an enrichment of transitions from the RepActPro state into the activating states (ActGene and ActProm) on mesenchymal and chondrogenic genes. On mesenchymal genes, we observed small but directed effects from ESC to the more differentiated bmMSC, which are expected to express mesenchymal genes. On chondrogenic genes, the enrichment of this transition is more prominent and also clearly directed, with the effect size increasing with the differentiation from ESC to bmCC (Fig. 4). Importantly, the reverse transition, the gain of H3K27me3 on active promoters to form the RepActPro state, is not detectable in the ECL data.
The opposite was true for the MCL data, in which the RepActPro state rarely switched into activating promoter states. Instead, we found an enrichment of transitions from the activating promoter states into the RepActPro state on mesenchymal and chondrogenic genes. Importantly, although the effect sizes were relatively weak, the effect was clearly directed. On mesenchymal genes, the transition became more pronounced with increasing developmental distance from MSC to PC and MSC to HC, reflecting the repression of mesenchymal genes in mature PC and HC. On chondrogenic genes, the transition was mainly found with the differentiation into HC indicating that a subset of chondrogenic genes was downregulated with the onset of hypertrophy (Fig. 4).
To test the hypothesized critical association of the RepActPro state and downregulated gene expression, we used the extensive published knowledge about gene expression changes during chondrogenesis and inspected the genes undergoing the transition into the RepActPro state in detail. For the mesenchymal genes, this transition was detected on genes like Pdgfra, Itga3, and Fzd1 (Supp.Fig. B13A), which are downregulated upon the commitment to the chondrogenic fate [6, 26, 34]. The chondrogenic genes were split into two subsets: One subset showed the gain of H3K27me3 from MSC to PC and HC and included genes like Tgfb1, Hoxa11, Shox2, Wnt10b, and Msx2, which are closely associated with the initiation of the chondrogenic fate in MSC, but are downregulated upon its establishment [7, 39, 47, 57, 72] (Supp.Fig. B13B, Supp.Fig. B7). In the second subset, the transition occurred between PC and HC. This subset included genes like Col2a1, Col27a1, Runx3, Pth1r, Wnt5b, Rflna, and Acan, which are expressed in PC but downregulated in HC [27, 37, 38, 43, 65, 73, 74] (Supp.Fig. B13C, Supp.Fig. B7). Together these data strongly support a model in which the addition and removal of H3K27me3 on promoters marked as active are tightly associated with alterations in gene expression, thereby contributing to the control of cell lineage differentiation.
Comments (0)