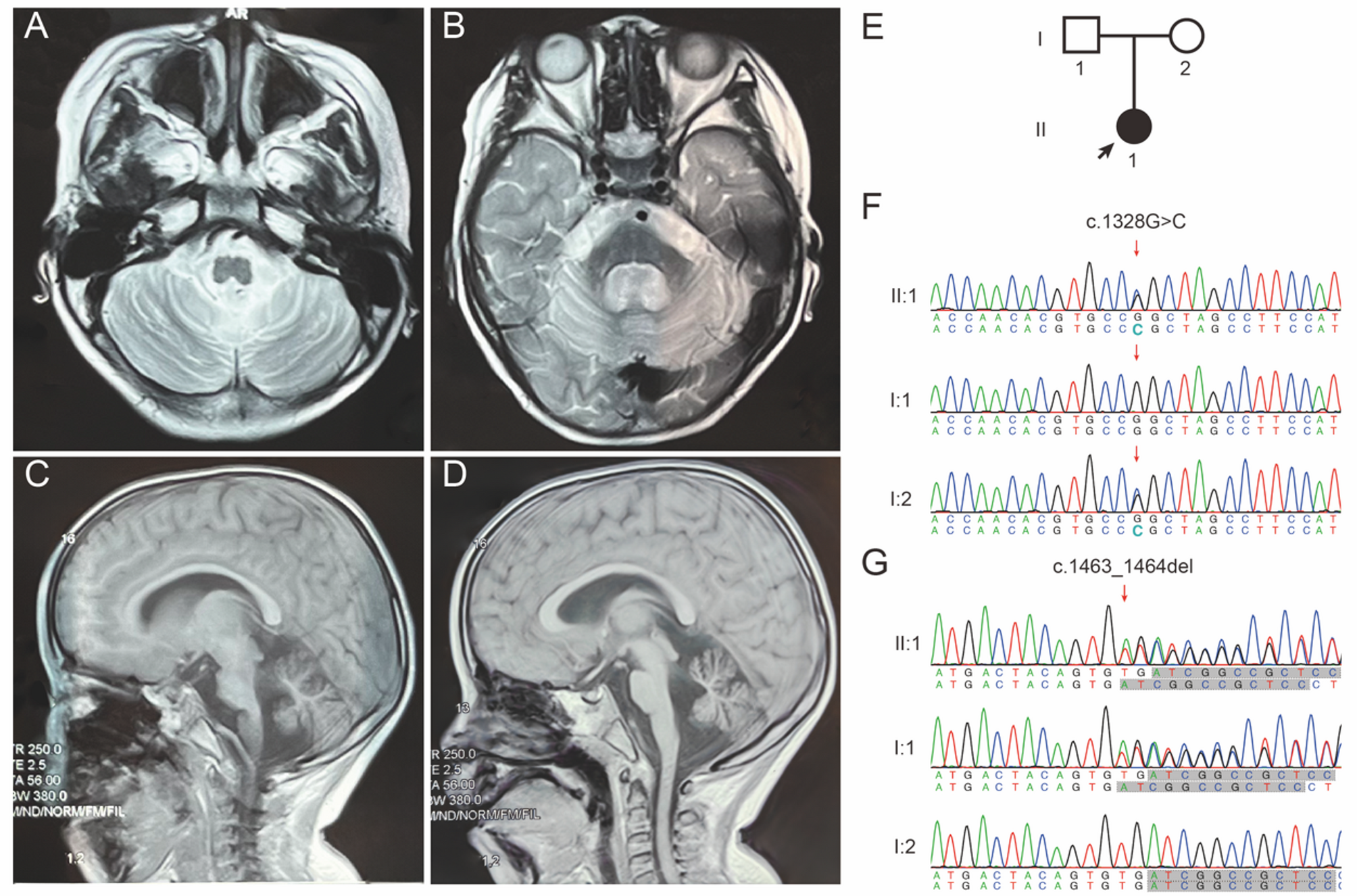
Our study introduces the first documented case of a child exhibiting intellectual disability, dysarthria characterized by a unique singsong speech pattern, cerebellar atrophy, and consistently elevated total plasma cholesterol levels. WES has identified that she carries compound heterozygous variants in the EEFSEC gene. Our findings are consistent with extensive genetic research that underscores the essential role of selenoprotein synthesis in human health and development. The clinical manifestations observed in this patient offer unprecedented insights into the phenotypic range related to disturbances in selenoprotein biosynthesis, with a specific emphasis on the involvement of the EEFSEC gene.
The synthesis of selenoproteins is a complex, multi-step process essential for the proper integration and functionality of selenium, an indispensable micronutrient, in its selenocysteine form (Sec) [17]. This form is acknowledged as the 21 st proteinogenic amino acid. The emergence of developmental delays, neurological impairments, and oxidative stress due to variants in this biosynthesis pathway’s components highlights the critical nature of these processes [17, 18]. As our patient exhibits compromised development potentially linked to EEFSEC gene variants, eEFSec protein has been shown to specifically bind the Sec-tRNA[Ser]Sec. It is hypothesized to operate autonomously from a guanine nucleotide exchange factor, owing to its high intrinsic GTP affinity [5, 6]. Consequently, the EEFSEC gene plays a vital role in decoding UGA codons as selenocysteine, rather than as termination signals.
The protein expression assay revealed a significant reduction in the expression level of the R443P variant compared to the wild-type. Human eEFSec protein is structured into four domains (D1-D4), creating a chalice-like formation. Detailed analysis of the D3 domain uncovers multiple loop insertions prone to solvent exposure, notably between β17 and β18 (residues 352–373), β21 and β22 (residues 432–444), and β18 and β19 (residues 378–410). The R443P missense variant is situated within the D3 domain, a region responsible for maintaining eEFSec protein stability. Research indicates that the D4 domain’s orientation is solidified through interactions between the loop β28-α11 at the extreme C-terminus and D3 residues. The formation of a hydrogen bond between Glu372 in D3 and Lys582 in the C-terminal segment is particularly pivotal [6]. The K582A and 582KRYVF586 -> AAAAA variants show reduced expression levels and stability compared to wild-type eEFSec [6]. This underscores the significance of amino acid residue hydrogen bonds in maintaining eEFSec’s structural integrity. We therefore propose that the R443P variant diminishes amino acid interactions at position 443 and disrupts key hydrogen bonds, potentially destabilizing eEFSec’s chalice-like structure. This structural perturbation likely reduces eEFSec stability, ultimately impairing its functional activity. On the other hand, although the V488Dfs*113 variant does not exhibit significant changes in protein expression, it induces structural alterations in the D4 and Linker domains. Previous studies have shown that these regions are critical for binding to the variable stem and loop of tRNASec, while the D4 domain specifically interacts with the apical loop of SECIS — both essential for UAG codon decoding. Variants or deletions in D4 have been demonstrated to severely impair eEFSec activity and selenoprotein synthesis. Therefore, the V488Dfs*113 variant is likely to disrupt eEFSec function. The decreased SELENOF levels and GSH activity in our patients support the conclusion that the EEFSEC compound heterozygous mutations impair eEFSec’s efficiency in translating the UGA codon, thereby hindering adequate selenoprotein synthesis.
Selenoproteins are known for their antioxidant properties, effectively neutralizing reactive oxygen species (ROS) [18]. While ROS play a crucial role in various biological functions such as cellular communication, growth modulation, oncogenesis inhibition, and immune defense at moderate levels, an overabundance can lead to oxidative stress, causing harmful alterations to DNA, proteins, and lipids [19, 20]. This, in turn, triggers a range of diseases, particularly those associated with neuronal degeneration. Our study expands the knowledge of lipid peroxidation products in the context of disorders of selenoprotein synthesis. Increased levels of oxidized fatty acid metabolites in the patient’s plasma, compared to healthy individuals, shed light on an aspect of oxidative stress potentially pivotal in the pathogenesis of conditions related to EEFSEC variants. This finding is consistent with previous research on an individual with variants in the SBP2 gene [21] and offers a biochemical basis for the phenotypic symptoms observed in our patient. Although we detected an increase in the patient’s cholesterol levels, we were unable to measure 7β-hydroxycholesterol, which might better explain the observed rise in cholesterol. Additionally, the observed reductions in multiple oxylipins, including 5(6)-DiHET, 16(17)-DiHDPA, and some HDHA/HEPE species, suggest a broad suppression of polyunsaturated fatty acid (PUFA) metabolic pathways. This systemic alteration in lipid mediator networks may potentially impact neurodevelopment [22, 23], although the precise relationship between PUFA metabolism and selenoprotein pathways requires further elucidation. The underlying mechanisms driving these metabolic changes remain to be fully investigated.
SEPSECS, which encodes the O-phosphoseryl-tRNA: selenocysteinyl-tRNA synthase, is upstream in the selenoprotein synthesis pathway where eEFSec is located and has been associated with PCCA [10, 11]. Patients compound heterozygous for SEPSECS variants c.974 C > G (p.Thr325Ser) and c.1287 C > A (p.Tyr429*) exhibit reduced levels of selenocysteinyl-tRNA in brain tissue and decreased expression of three selenoproteins: thioredoxin reductase TXNRD1 and glutathione peroxidases GPX1 and GPX4 [10]. This finding parallels the observed reduction in GSH activity in our patient. Patients with SEPSECS variants typically present with neonatal irritability, spastic or dystonic quadriplegia, virtually absent psychomotor development, axonal neuropathy, and elevated blood/CSF lactate. Neuropathological examination reveals laminar necrosis, severe myelin loss, neuron depletion, and astrogliosis. In contrast, our patient’s primary features are motor developmental delay, inability to sit or walk independently, dysarthria, normal muscle tone, positive pathological signs, low mood, lack of vitality, normal blood lactate levels but recurrently elevated total cholesterol. A common finding in imaging studies is cerebellar atrophy [10]. Whether due to a synthesis block of selenocysteinyl-tRNA caused by SEPSECS gene variants or a translation impediment of selenoprotein UGA codon caused by EEFSEC gene variants, both lead to cerebellar atrophy in patients, underscoring the importance of selenoproteins in maintaining cerebellar function. Additionally, the elevated oxidative stress markers in our patient’s plasma suggest that cerebellar atrophy may be a consequence of an imbalance in oxidative stress.
In fact, disorders in selenoprotein synthesis can lead to the involvement of multiple systems and present with diverse symptoms. For instance, variants in the SBP2 gene, which is also involved in the translation of the selenoprotein UGA codon, can lead to characteristic clinical manifestations and the implications of growth hormone (GH) and triiodothyronine in longitudinal bone growth and maturation [24, 25], cutaneous photosensitivity and enhanced UV-mediated oxidative damage, azoospermia with spermatogenic maturation arrest, axial muscular dystrophy similar to SEPN1-related myopathies, impaired T cell proliferation and abnormal cytokine production, increased fat mass but enhanced insulin sensitivity, and hearing loss [12]. However, our patient’s thyroid function is normal, and to date, there has been no detection of photosensitivity, immune disorders, or hearing loss, highlighting the clinical phenotype complexity associated with selenoprotein-related diseases. Moreover, certain genetic polymorphisms in the EEFSEC gene have been identified as potential risk factors for various diseases, including chronic obstructive pulmonary disease [26], preterm birth [27, 28], and periodontitis [29, 30]. However, no increased susceptibility to these conditions was observed in either the patient or their family members. This phenomenon may be attributed to variations in selenium-protein deficiency susceptibility stemming from the patient’s unique environmental factors and genetic background.
Given the essential role of selenium and selenoproteins in various biological processes, variants in genes involved in selenoprotein biosynthesis, such as eEFSec, can have profound implications. These implications extend beyond oxidative stress to include neurological development [31, 32], immunity [33,34,35], and thyroid hormone metabolism [25], among others. Consequently, there’s a pressing need for in-depth research into the pathophysiological mechanisms of disorders tied to selenoprotein synthesis, particularly concerning the EEFSEC gene. Such research could provide critical insights into developing targeted therapeutic strategies for affected individuals.
In summary, we report a child with progressive cerebellar atrophy caused by variants in the EEFSEC gene and confirm its role in the pathogenesis of the disease in the reported case. Our findings emphasize the importance of considering EEFSEC gene variants in patients presenting with similar clinical spectrums. Progressive cerebellar atrophy due to a compound heterozygous pathogenic variant in the EEFSEC gene expands the genetic subtypes of this disease. Variants in genes involved in selenoprotein translation, such as SEPSECS and EEFSEC, can lead to progressive cerebellar atrophy, demonstrating the vital role of selenoproteins in maintaining cerebellar function.


Comments (0)