
Remember me



You have full access to this article via your institution.
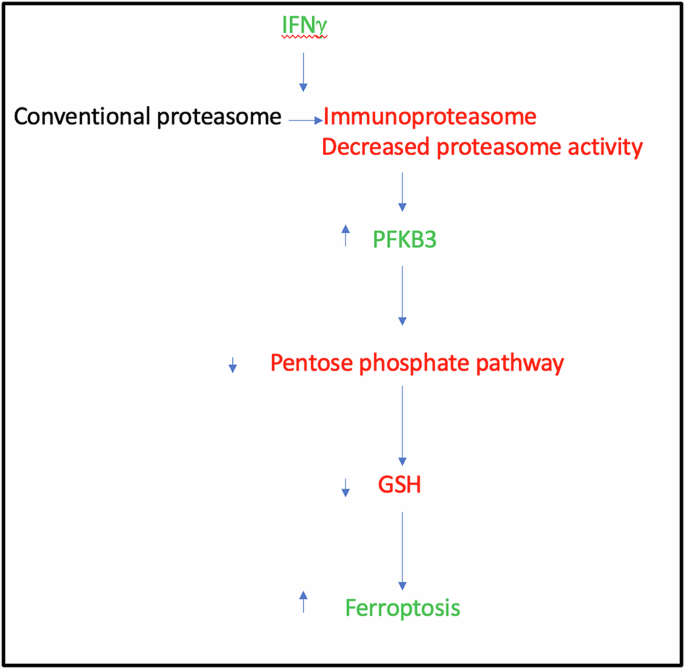
Woo et al. reported in Cell that in multiple sclerosis (MS) mouse models and likely in human MS, a cascade is initiated by neuroinflammation, which leads to a switch in neurons from the conventional proteasome to the immunoproteasome, compromising proteasome activity. This results in elevation of PFKB3, which lowers antioxidant defenses, making neurons more susceptible to ferroptotic cell death.
Multiple sclerosis (MS) is a chronic, debilitating central nervous system (CNS) disease that is immune-mediated. MS typically presents between the ages of 20 and 30 years and can follow different clinical courses. Most common is the relapsing-remitting form of the disease, where episodes of dysfunction are interspersed with periods of remission with at least some recovery, followed by subsequent relapses. When remissions stop, it becomes primary progressive MS, which is characterized by a steady functional decline without remissions. Occasionally, MS can present with an aggressive demyelinating disease.
Researchers have modeled human MS by immunizing mice with myelin antigens, which provoke an autoimmune response, to establish the experimental autoimmune encephalomyelitis (EAE) models. Despite the differences between the EAE approach using exogenous antigens and human MS involving autoantigens, EAE models have supported the development of a number of drugs for MS, although many drugs showing benefits in these mouse models have ultimately failed in humans.1 EAE experiments on standard mouse genetic backgrounds like C57BL/6 result in a monophasic and progressive disease. To capture the relapsing-remitting form of MS, researchers have also used the Biozzi ABH mouse, which has a reproducible relapsing-remitting course.2
In MS, neuroinflammation is accompanied by demyelination and gray matter neurodegeneration. The neurodegenerative component appears to be a critical determinant of functional outcome, underscoring the importance of understanding this process.3 One pathway that has received some attention in MS is ferroptosis, a form of non-apoptotic cell death mediated by overwhelming lipid peroxidation of phospholipids in cell membranes. Ultimately, this phospholipid modification makes the plasma membrane stiffer and prone to fracture, which leads to subsequent cell death.4 In MS, there is increased lipid peroxidation and iron deposition around the rims of inflammatory lesions, since iron is an important driver of lipid peroxidation. Furthermore, there is decreased expression of GPX4, a key enzyme that buffers ferroptosis by converting lipid hydroperoxides into lipid alcohols. In experimental models of MS, there appear to be benefits of anti-ferroptosis drugs.4,5
An important theme in neurodegenerative disease is the failure of protein degradation processes like autophagy and the ubiquitin-proteasome pathway. The proteasome is a barrel-shaped complex that degrades ubiquitinated, unfolded, and monomeric proteins into peptides. It has two components. The 20S core has the catalytic activities, and the 19S cap is involved in functions like substrate deubiquitination and opening of the entry “gate” of the 20S proteasome. The 20S has chymotrypsin-like, trypsin-like, and peptidyl-glutamyl-peptide hydrolyzing activities associated with the β1c (PSMB1), β2c (PSMB2) and β5c (PSMB5) subunits, respectively. In response to IFNγ signaling in many cell types, these components are replaced by immunoproteasome subunits β1i (PSMB9), β2i (PSMB10) and β5i (PSMB8) subunits, respectively. This response enables cells to present a diverse repertoire of peptide antigens on MHC class I to facilitate immune responses. This switch is generally associated with increased proteasome activity. However, the consequences of the switch to the immunoproteasome in neurons exposed to such inflammatory stimuli are poorly understood.
Woo et al.6 have recently made a series of important discoveries that link inflammation, immunoproteasome and ferroptosis in MS pathogenesis (Fig.1). They observed that the immunoproteasome component, PSMB8, is expressed in neurons and that its levels are elevated in cultured neurons after exposure to IFNγ, in neurons in active human MS, and in EAE and Biozzi ABH mouse models, while the levels of its conventional proteasome counterpart, PSMB5, are unchanged. Importantly, they suggested that the elevation of PSMB8 (immunoproteasome) drives neurodegeneration in the EAE model, as this could be rescued by an immunoproteasome inhibitor. Furthermore, in mice with pan-neuronal knockout of PSMB8, there was a rescue of neuronal loss but no differences from EAE-treated wild-type mice with regard to microglial activation, CNS-infiltrating T cells, and IL-17 and IFNγ-producing T cells. Furthermore, PSMB8 overexpression was sufficient to cause neurodegeneration, which was not seen with overexpression of its conventional proteasome counterpart, PSMB5.
Fig. 1: Schematic of a cascade of events induced by neuroinflammation in MS models.
IFNγ causes a switch from the conventional proteasome to the immunoproteasome in neurons in MS models, which leads to a decrease in proteasome activity, resulting in the accumulation of the proteasome substrate PFKB3. This decreases the activity of the pentose phosphate pathway, which in turn lowers GSH levels and renders neurons susceptible to ferroptosis.
Neuronal PSMB8 overexpression was associated with fewer transcripts related to synaptic function and induction of genes involved in redox balance. Indeed, the authors then showed higher levels of ROS in PSMB8-overexpressing but not in PSMB5-overexpressing neurons. They found that PSMB8 overexpression decreased overall proteasome activity in neurons (in contrast to what was previously seen in astrocytes, microglia, and cell lines). EAE mice had impaired proteasome activity (increased K48-linked ubiquitinated proteins), which was ameliorated by PSMB8 deletion. These data and others in this comprehensive study showed that inflammatory processes in MS models and likely in human MS lead to a switch to immunoproteasomes in neurons, which compromises proteasome activity.
Remarkably, many of the toxic effects of proteasome inhibition in this setting could be attributed to elevation of the proteasome substrate, phosphofructo-2-kinase/fructose-2,3-bisphosphatase (PFKB3). In neurons, PFKB3 activates glycolysis and inhibits the pentose phosphate pathway. In cultured neurons, PFKB3 levels were increased after PSMB8 overexpression, and chronic IFNγ treatment increased PFKB3 expression, an effect that was abrogated by PSMB8 knockout. Likewise, PFKB3 levels were elevated in different EAE scenarios in mice and human MS (both in lesions and apparently healthy-looking regions). Genetic deletion of PSMB8 ameliorated the PFKB3 elevation in EAE mice.
The authors then used a range of approaches to show that the elevation of PSMB8 results in the expected downregulation of the pentose phosphate pathway and lowered levels of NADPH, which is crucial to maintaining levels of the antioxidant glutathione (GSH). GSH is a key substrate for the ferroptosis buffer, GPX4. Indeed, PSMB8- and PFKB3-overexpressing neurons exposed to glutamate (which activates ferroptosis independently of effects on glutamate receptors) or a GPX4 inhibitor had higher levels of pro-ferroptotic lipid peroxidation. In mice, the elevated levels of lipid peroxides induced by EAE were attenuated by neuronal PSMB8 knockout. Similarly, treatment of mice with a PFKB3 inhibitor (pfk-158) or neuronal PFKB3 knockout ameliorated neurodegeneration in EAE-treated mice. Importantly, these benefits appear to be downstream of the effects of EAE on the proteasome, neuroinflammation, and demyelination.
In summary, Woo et al. show that in EAE mouse models and likely in human MS, there is a cascade that is initiated by neuroinflammation, which leads to a switch in neurons to the immunoproteasome, which compromises the proteasome activity.6 This results in elevation of PFKB3, which lowers antioxidant defenses, making the neurons more susceptible to ferroptosis. This is an interesting and important study for a number of reasons. From an MS perspective, it highlights the role of ferroptosis in neuronal cell death and introduces new therapeutic targets for further consideration. Ferroptosis has been associated with MS in previous studies and some of them have shown benefits of anti-ferroptosis strategies in mouse models. However, much of the previous attention has been on ferroptosis in glial cells/oligodendrocytes. In a broader perspective, this work raises the possibility that the pathway from IFNγ to immunoproteasome activation/proteasome inhibition to ferroptosis in neurons may operate in other contexts, too.
Comments (0)