
Remember me



You have full access to this article via your institution.
Rather than a simple mode of cell death, β-cell failure in type 2 diabetes is a crisis of identity. Zinc is profoundly important and indispensable for the processing and storage of insulin; however, a study by Ma et al. shows that zinc accumulation triggers an integrated stress response and initiates the conversion of β-cells into α-like cells through a zinc-ATF4-ARX regulatory axis.
Zinc (Zn2+), a tiny metal ion stored in the insulin granules of β-cells at concentrations 1000-fold higher than in other cell types, is a determinant of β-cell identity, function, and survival.1 A central component of the insulin hexamer that enables the crystallization and stabilization of insulin, Zn2+ is secreted with insulin and subsequently re-imported into the cell. Extensive Zn2+ trafficking occurs across the β-cell membrane, and intra- and extracellular Zn2+ concentrations are important for glucose homeostasis and β-cell development. These characteristics make Zn2+ an attractive tool for β-cell-specific therapy, as Zn-based prodrug systems can deliver small molecules selectively into β-cells.2
However, disturbances occur within this tightly regulated physiological system. An extreme shift occurs during early β-cell development, when Zn2+ is essential for RNA synthesis and organelle formation. Zn2+ accumulates to high levels within β-cells, coinciding with an increased number of insulin secretory granules and increased expression of zinc transporter 8 (ZnT8). ZnT8 is expressed almost exclusively in pancreatic islets and serves as the primary mediator of β-cell Zn2+ enrichment. After final maturation, granules are released during glucose stimulation, resulting in increased extracellular Zn2+ levels.
Indispensable for numerous physiological processes, Zn2+ supports not only insulin stability but also the activity of antioxidant enzymes, including superoxide dismutase, catalase, and glutathione peroxidase, which are expressed in β-cells at relatively low levels.
Logically, one should therefore “think zinc”3 and take dietary Zn2+ supplements, which are widely available on the market. Indeed, Zn2+ supplementation has been shown to enhance β-cell function and viability under physiological Zn2+ levels. However, higher doses cause toxicity. Randomized trials have reported modest improvements in glycemic endpoints among individuals with established type 2 diabetes (T2D); however, overall evidence remains insufficient to support Zn2+ supplementation in healthy or pre-diabetic populations.4
Zn2+ is a double-edged sword. What is beneficial in balance is harmful in excess; dysregulation of Zn2+ homeostasis is increasingly recognized as a contributing factor in both type 1 diabetes (T1D) and T2D. Excessive Zn2+ may disrupt both metabolic and hormonal homeostasis.4 Paradoxical findings have been reported regarding Zn2+ cellular localization, paracrine mechanisms within islets, and mutations affecting the β-cell-specific ZnT8 (SLC30A8), which mediates zinc transport from the cytosol into secretory granules5 and is also an autoantigen in T1D. In mice, loss-of-function mutations in Slc30a8 impair insulin crystallization, granule formation, and β-cell function, although marked differences exist between strains.3
In apparent contrast, SLC30A8 insufficiency in humans protects against T2D in both heterozygous6 and homozygous7 carriers. In human β-cells, loss of SLC30A8 function improves cell maturation and function, prevents lipo- and glucotoxicity-induced cell death, and prevents endoplasmic reticulum stress, all through reduction of intracellular zinc levels.8,9,10 Why the loss of a gene product that is strongly expressed in adult β-cells and required for normal insulin crystallization confers protection against T2D remains unclear.
In support of the hypothesis that lower islet Zn2+ levels protect against T2D, several T2D-associated ZnT8 variants are associated with excessive islet zinc levels, whereas distinct ZnT8 variants with reduced β-cell Zn2+ levels are associated with significantly lower T2D risk.
Zn2+ has also been proposed to participate in the fine-tuning of islet hormone crosstalk by affecting glucagon secretion from neighboring α-cells and by suppressing insulin secretion through a negative feedback loop. However, several contradictory results have been reported, and no precise mechanistic investigations of a direct effect of Zn2+ on glucagon secretion have been conducted.5
Ma et al. add another level to the Zn2+ duality in the context of T2D; they show that Zn2+ triggers β-to-α-cell transdifferentiation (Fig. 1). In islets from patients with T2D, they found substantially higher Zn2+ levels and ZnT8 expression in β-cells, particularly in bi-hormonal insulin+/glucagon+ cells. Re-analysis of human islet single-cell RNA sequencing (scRNA-seq) data from the Human Pancreas Analysis Program database revealed a shift in endocrine cell composition in T2D compared with non-diabetic individuals, with an increase in α-cells, a reduction in β-cells, and elevated ZnT8 expression.
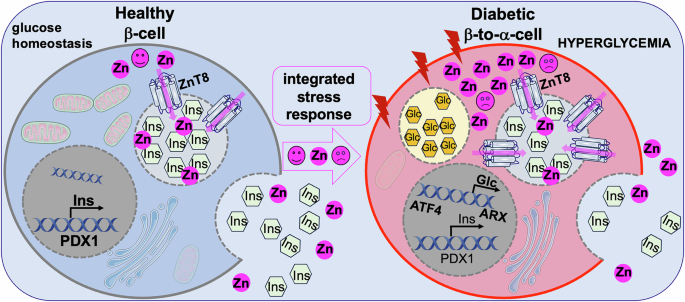
Fig. 1: Dual actions of Zn2+ in pancreatic β-cell function and fate.
Under physiological conditions (left), Zn2+ is transported into insulin secretory granules by ZnT8, where it supports insulin crystallization, granule integrity, and regulated insulin secretion, thereby promoting β-cell function and survival. In contrast, under diabetic or stress conditions (right), dysregulated zinc homeostasis leads to excessive intracellular Zn²+ accumulation and altered subcellular distribution. Elevated Zn2+ levels activate cellular stress pathways, including the integrated stress response, resulting in ATF4 activation and induction of ARX, a transcription factor that promotes α-cell identity. This transition promotes loss of β-cell identity, impaired insulin secretion, and ultimately β-to-α-cell transdifferentiation. (Figure created using smart servier medical art under https://creativecommons.org/licenses/by/3.0/).
Similarly, in diabetic mice fed a high-fat diet, the emergence of bi-hormonal insulin+/glucagon+ cells and the occurrence of β-to-α-cell transdifferentiation both correlated with increased ZnT8 expression and higher intra-islet zinc levels. Genetic upregulation of ZnT8 induced loss of β-cell identity; bi-hormonal cells exhibited high ZnT8 expression, indicating that elevated ZnT8 expression is the driver of β-cell identity loss. To further test causality, the authors increased intracellular Zn2+ levels in multiple systems: human islets, stem cell (SC)-derived islets, a mouse model of T2D induced by a high-fat diet, and a mouse islet transplantation model. Zinc supplementation increased ZnT8 expression and intracellular zinc levels in β-cells, reduced β-cell and increased α-cell markers, increased the number of bi-hormonal cells, and reduced glycemic control. Genetic lineage tracing showed that cells originally expressing β-cell markers began to express glucagon, indicative of transdifferentiation. Collectively, these results show that Zn2+ accumulation within β-cells is not merely correlative but is sufficient to induce identity changes under diabetic stress.
ScRNA-seq analyses revealed that excessive Zn2+ activates the integrated stress response (ISR), which is characterized by elevated ATF4, a transcription factor downstream of eIF2α phosphorylation that is central to ISR signaling. ATF4 induction correlates with increased expression of ARX, a master regulator of α-cell identity, suggesting a direct transcriptional mechanism for β-to-α lineage conversion.
Importantly, Ma et al. targeted Zn2+ accumulation as a therapeutic strategy to preserve β-cell identity and function by ZnT8 genetic depletion or pharmacological inhibition using SC-derived islets transplanted into hyperglycemic mice. The antibiotic anisomycin, classically used as a global inhibitor of protein synthesis, was identified as a compound that suppresses ZnT8 translation at low nanomolar concentrations, thereby inhibiting intracellular zinc transport. Both approaches reduced intracellular zinc accumulation, prevented β-to-α-cell transdifferentiation, and improved glycemic control. These protective effects were reversed by Zn2+ supplementation, confirming that the effects of both interventions are specific to Zn2+. Notably, low-dose anisomycin significantly inhibited zinc transport without causing global translational arrest. This activity as a selective regulator of metal homeostasis represents a pharmacological breakthrough, suggesting that well-known but toxic compounds may have applications in precision medicine.
Several caveats remain. First, the levels of extracellular Zn2+ used experimentally may not fully reflect those that are present in vivo. Second, other studies (e.g., Sui et al.10) have not detected major changes in cellular identity in human ZnT8 knockout SC-derived β-cells as would be predicted from the present results. These apparent discrepancies require further investigation. Nevertheless, the study by Ma et al. identifies a novel mechanism of β-cell identity loss driven by Zn2+ accumulation under metabolic stress. Reducing Zn2+ accumulation via ZnT8 inhibition represents a promising strategy for preserving functional mature β-cells and improving glycemic control in T2D prevention and therapy.
Comments (0)