Patient recruitment
This study was conducted with ethical approval from the Luoyang Orthopedic Hospital of Henan Province and the Capital Medical University Xuanwu Hospital Ethics Committee. For RNA sequencing, paravertebral muscle tissues were collected from three individuals with relatively normal muscle condition and three with severe degeneration. Additionally, 10 patients diagnosed with PMD and 10 individuals without PMD were recruited for qRT-PCR validation. All participants provided written informed consent prior to participation. The cross-sectional area of muscle is a key parameter for assessing paravertebral muscle health, with a larger area indicating better muscle quality. We selected the L4/L5 segment of the lumbar spine for sample collection due to its relatively large average muscle cross-sectional area, ease of measurement, and its frequent use in paravertebral muscle research [15, 17, 21]. The severity of PMD was evaluated using the Goutallier grading system, which provides a visual, semi-quantitative assessment of fat infiltration [17, 21, 22]. Individuals with Goutallier grades 0–1 were classified as the normal group, while those with grade 4 were assigned to the severe PMD (SPMD) group.
Muscle digestion
The paravertebral muscle tissues were placed in sterile phosphate buffer saline supplemented with 4% streptomycin (Gibco Thermofish). The tissues were washed twice with the same solution to ensure cleanliness. The visible connective tissue, blood vessels, and adipose tissue were then carefully removed. The cleaned muscle samples were then enzymatically digested using a mixture of Collagenase II, Dispase II, and a red blood cell lysis reagent [15, 17].
RNA sequencing and data analysis
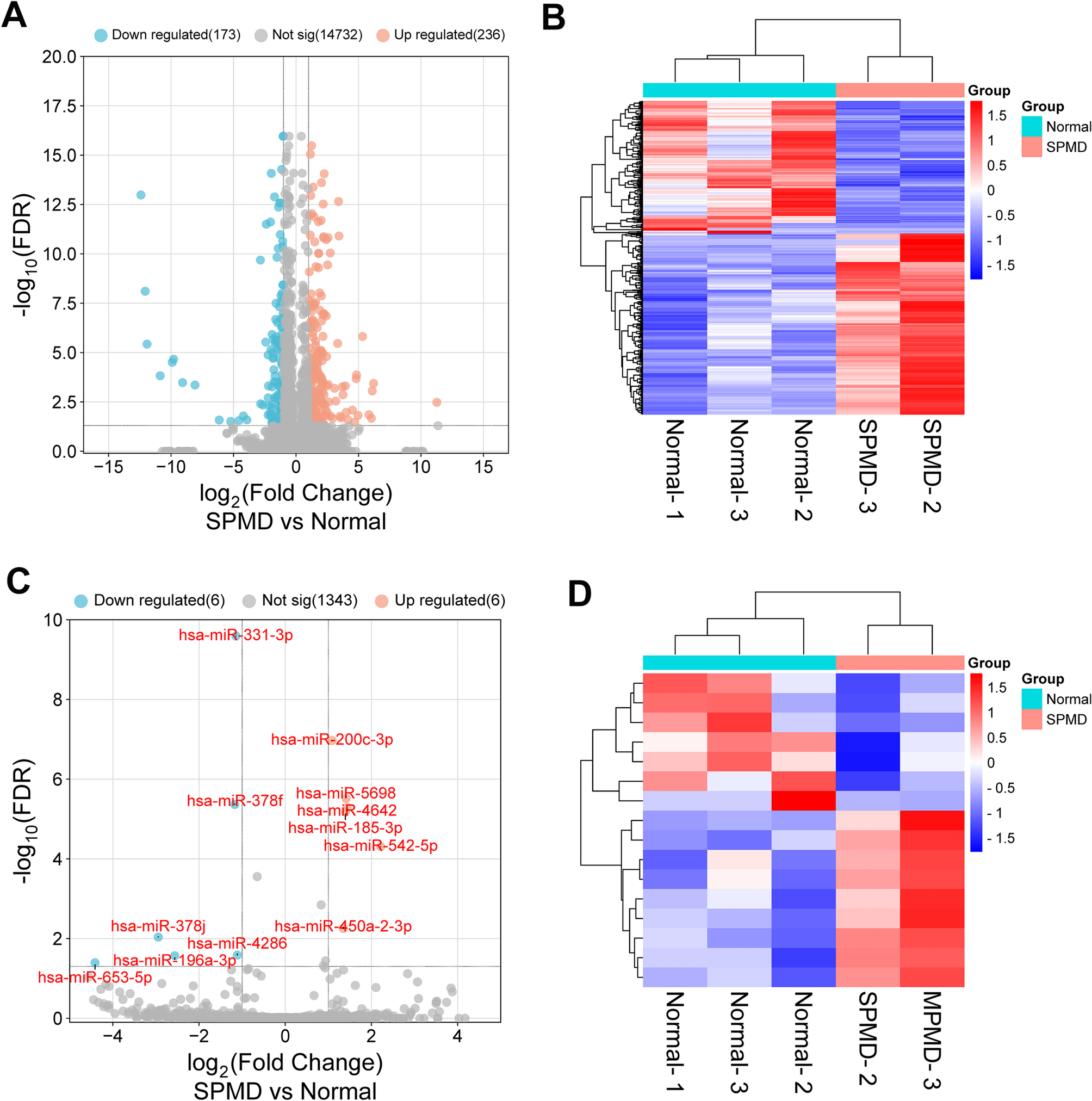
The total RNA was extracted from adult paravertebral muscle tissues using the TRIzol reagent (Life Technologies, USA). The purity and integrity of RNA were assessed using a NanoPhotometer spectrophotometer and an Agilent 2100 bioanalyzer. RNA library preparation was carried out following the manufacturer’s instructionsusing the NEBNext® Ultra™ RNA Library Prep Kit. RNA sequencing was performed by NovelBio Biopharmaceutical Technology Company. After data quality control, the gene expression was quantified using FPKM values with a detection threshold of FPKM ≥ 0.5 in ≥ 70% of samples per group; miRNA expression required normalized counts ≥ 5 in ≥ 50% of samples. The differentially expressed genes (DEGs) and differentially expressed miRNAs (DEMs) were analyzed using R software EBSeq package with statistical significance defined as|log2 fold-change (FC)| >1 and false discovery rate (FDR) < 0.05. Data visualization, including cluster heatmaps, volcano plots, and Sankey diagrams were output using R software pheatmap package, enhancedVolcano package, and networkD3 package, respectively [23].
Identification of the functional DEGs in PMD
Gene sets related to the extracellular matrix, oxidative stress, and ferroptosis were obtained from studies by Li et al. [19, 20], while genes associated with. autophagy, cell senescence, inflammasomes, pyroptosis, and apoptosis were sourced from Qiu et al. [24]. These genes were listed in Supplementary Table S1. To identify the functional DEGs associated with PMD, Venn plots were used to merge these genes with DEGs obtained from RNA-seq to screen for overlapping genes.
Protein-protein interaction (PPI) analysis
Search tool for recurring instances of neighbouring genes (STRING) database (https://string-db.org/) was used to build a PPI network for functional differentially expressed genes [25]. First, the DEGs were entered into the STRING database to obtain the interaction network and associated data files. These data were then imported into the Cytoscape software for visualization and further analysis. Key hub genes were subsequently identified using five algorithms (maximum clique centrality (MCC), Degree, Betweenness, EPC, and Closeness) available through the CytoHubba in Cytoscape [26]. Genes consistently ranked in the top 10 by all methods were defined as high-confidence hubs.
Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analysis
GO resource (http://geneontology.org) is an internationally standardized gene function classification database, encompassing three major categories: biological processes (BP), cell components (CC), and molecular functions (MF). It provides structured, computable information about gene and gene product functions [27]. KEGG (https://www.genome.jp/kegg/) pathway plays an crucial role in the gene annotation process, helping to identify the signaling pathways in which genes are involved [28]. The cutoff was defined as p-value < 0.05. BP and KEGG can directly reflect the potential function of genes. We used the R software clusterProfiler package (v4.0) for GO and KEGG enrichment analysis.
Construction of the DEMs-hub DEGs signaling network in PMD
For the key functional hub DEGs identified above, the upstream miRNAs of them were predicted using mirDIP 5.2 version (https://ophid.utoronto.ca/mirDIP/index.jsp) database [29]. PMD-related DEMs were then determined by intersecting the DEMs obtained from RNA sequencing with the predicted upstream miRNAs of each hub DEG from mirDIP. Based on these results, a comprehensive DEM–hub DEG regulatory network was constructed using Cytoscape software [26].
Transcription factor (TF)-gene regulatory network construction
NetworkAnalyst 3.0 database (https://www.networkanalyst.ca/) is a visual analytics tool for comprehensive gene expression profiling [30]. TF–target gene interaction data were sourced from the JASPAR database (http://jaspar.genereg.net/), which is integrated into the NetworkAnalyst 3.0 platform [31]. The TF-gene co-regulatory network was constructed in NetworkAnalyst 3.0 by uploading the top 10 hub DEGs (|log2FC| >1, p < 0.05) into the gene list input field with ‘Homo sapiens’ selected as the species. JASPAR database (TF-motif predictions, p < 1 × 10-⁵) was used under ‘TF-gene interactions’, and nodes were filtered by Degree > 1 and Betweeness > 0 to prioritize biologically relevant connections. The resulting network was then visualized using Cytoscape software [26].
Predicting the prognostic value of the key hub DEGs and DEMs
Receiver Operating Characteristic (ROC) curve and Precision Recall (PR) curve are commonly used to predict accuracy. In this study, these analyses were performed using the pROC package and ggplot2 packages in R software. The diagnostic potential of key DEGs was evaluated based on the area under the curve (AUC), with values closer to 1, indicating stronger predictive performance.
Extraction and cultivation of primary muscle stem cells (MuSCs)
The MuSCs were isolated enzymatically based on the instructions described [32]. Briefly, human paravertebral muscle were obtained from lumbar disc herniation patients who undergo spinal surgery. The muscle was then mechanically disrupted into pieces using sterile scissors, and followed by digesting with 0.03 g of 0.2% collagenase II (Worthington, LS004176) supplemented with 4% penicillin‒streptomycin and 15 ml DMEM medium. This was followed by a second digestion step using an additional 15 mL of DMEM (Gibco, Cat# 11965092), supplemented with 0.04 g of 0.2% collagenase II, 2 ml of trypsin, 2 ml of DNase (1 mg/ml), and 1% penicillin-streptomycin were added to the digestion mixture for further digestion. After enzymatic digestion, the cell suspension was filtered through a 300-mesh nylon filter, and centrifuged at 350 × g for 5 min. The isolated MuSCs were then cultured in Ham’s F10 medium (CellWorld, C1662-875) supplemented with 20% fetal bovine serum (FBS), 1% penicillin‒streptomycin (Sigma, P4333), and 5 ng/ml basic fibroblast growth factor (PeproTech #100-18B) [33]. The culture dishes were coated with Matrigel (Corning, 354230). MuSCs cultures were placed in 37 °C incubators with 5% CO2.
RNA extraction and quantitative real-time RT-PCR
We used the TRlzol Reagent (Life Technologies, USA) to extract total RNAs from paravertebral muscle tissues. RNA purity and concentration were assessed using a microspectrophotometer (Allsheng, China), while RNA integrity was confirmed via gel electrophoresis. Complementary DNA (cDNA) was synthesized from the extracted RNA using the one Step PrimeScript™RT-PCR Kit (Takara, Japan). qRT-PCR was then carried out using the Thermal Cycler Dice™ Real-Time System III (Takara, Japan). After the qRT-PCR reaction, both melting curves and amplification curve were examined to confirm the specificity and efficiency of the amplification. The forward primers and reverse primers of each gene used in this study are listed in Supplementary Table S2. Their relative expression levels were measured using the 2−△△Ct method, as described previously [14, 19].
Western blotting
Total protein was extracted from normal and degenerative paravertebral muscle tissues using radioimmunoprecipitation assay (RIPA) lysis buffer along with a total protein extraction kit (Bestbio, Shanghai, China; Catalog #BB-3101) following the manufacturer’s protocol. The protein concentration was measured using the Micro Bicinchoninic Acid Protein (BCA) Assay Kit (Beyotime, China), which allows for accurate quantification of total protein content. Proteins were separated by Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis (SDS-PAGE) and subsequently transferred onto a Polyvinylidene Difluoride (PVDF) membrane (Merck, Germany). These membranes were incubated overnight at 4 °C with primary antibodies anti-ATF3 (ab207434, Abcam), anti-Aggrecan (ab3778, Abcam), anti-CDKN1A/P21 (ab102013, Abcam), anti-GPX4 (ab125066, Abcam), anti-Caspase3 (ab32351, Abcam), anti-GSDME (ab215191, Abcam), and β-actin (Proteintech, 66009-1-1 g) in 5% non-fat dry milk in PBS containing 0.1% Tween-20. The PVDF membranes were washed and incubated with the appropriate secondary antibody for 1 h at room temperature. Protein bands were detected using enhanced chemiluminescence (ECL) reagents (Beyotime, Shanghai, China) and visualized with the ChemiDoc™Touch Imaging System (Bio-Rad, Berkeley, CA, USA).
Detection of cell death
Cell death rates was measured by conducting flow cytometry analysis of cells stained with Annexin V and propidium iodide (PI). The VF647A-Annexin V/PI Apoptosis Detection Kit (HY-K1093-50T, MedChemExpress) was used to for this purpose. The treated cells were collected, washed with PBS, and stained with Annexin V-FITC and PI in the dark at room temperature for 15 min. The stained samples were then analyzed using a flow cytometer (BD FACSAriaTM II). Subsequently, FlowJo VX10 software was used to analyze the data. In flow cytometry apoptosis assays, Q2 typically indicates late apoptosis (Annexin V+/PI+), while Q3 indicates early apoptosis (Annexin V+/PI-). The sum of Q2 and Q3 is commonly used to quantify overall apoptosis.
Determination of glutathione (GSH), ROS, and fe2+ expression level
Based on the manufacturer’s protocols, GSH assay kit (CS0260, Sigma) and iron assay kit (Abcam, Cambridge, UK) were used to assess the relative GSH and iron concentrations in cell lysates. Intracellular ROS levels were detected using the oxidation-sensitive fluorescent probe DCFH-DA, following a previously described method. The analysis was performed with a ROS Assay Kit (Beyotime, China) and quantified via flow cytometry using the FACSCalibur system (BD Biosciences).
Enzyme-linked immunosorbent assay (ELISA)
Cell lysis were harvested following 24 h after transfection. According to the manufacturer’s protocols, human IL-1β and IL-6 ELISA kit (Elabscience, China) were used to measure the intracell concentrations of IL-1β and IL-6 under various treatment conditions. Briefly, 96-well plates were coated with 100 µL/well of capture antibody and incubated overnight at 4 °C. After washing (3× with wash buffer), plates were blocked with 300 µL/well of reagent diluent for 1 h at room temperature. IL-1β and IL-6 standards and normalized samples (50 µg total protein/well) were added in duplicate (100 µL/well) and incubated for 2 h at room temperature. Plates were incubated with detection antibody 1 h, and followed by streptavidin-HRP 30 min at room temperature. Absorbance was measured at 450 nm (correction at 570 nm) using a microplate reader (BioTek Synergy H1). IL-1β and IL-6 concentrations were calculated from standard curves using 4-parameter logistic regression.
Dual-Luciferase reporter assay
The binding site between EGR1 and ATF3 promoter was predicted using JASPAR database (https://jaspar.elixir.no/) [31]. pGL4.74[hRluc/TK] (Promega, backbone: Renilla luciferase) was used for normalization. The ATF3 promoter region (wild-type and mutant) was cloned into the pGL4.10[luc2] vector (Promega, backbone: Firefly luciferase). The ATF3 wild-type and mutant promoter were amplified by PCR from genomic DNA, the purified PCR products were ligated into the pGL4.10[luc2] vector and T4 DNA ligase. All constructs were verified by sanger sequencing. Full-length human EGR1 cDNA was subcloned into the pcDNA3.1(+) vector (Thermo Fisher Scientific) to overexpress EGR1. EGR1 siRNA was used to knockdown EGR1. The luciferase reporter vectors and EGR1 overexpression vector/siRNA were co-transfected to human embryonic kidney (HEK) 293T cells using Lipofectamine 8000 (Beyotime, China). Luciferase activity was measured using the Luciferase Assay Kit and GloMax® 20/20 Luminometer (Promega, Madison, WI, USA). Reporter gene activation was assessed by normalizing Firefly luciferase relative light units (RLU) to to Firefly luciferase RLU in each sample.
Statistical analysis
Statistical analysis of the quantitative data was performed using GraphPad Prism version 8. For data following a normal distribution, group comparisons were conducted using an unpaired Student’s t-test. If data violated normality assumptions, Mann-Whitney U test (unpaired) or Wilcoxon signed-rank test (paired) were used to analyze data. Results are expressed as mean ± standard deviation (SD). The P-value less than 0.05 represented statistical significance.* represents p < 0.05; ** represents p < 0.01 and *** represents p < 0.001.



Comments (0)