
Remember me
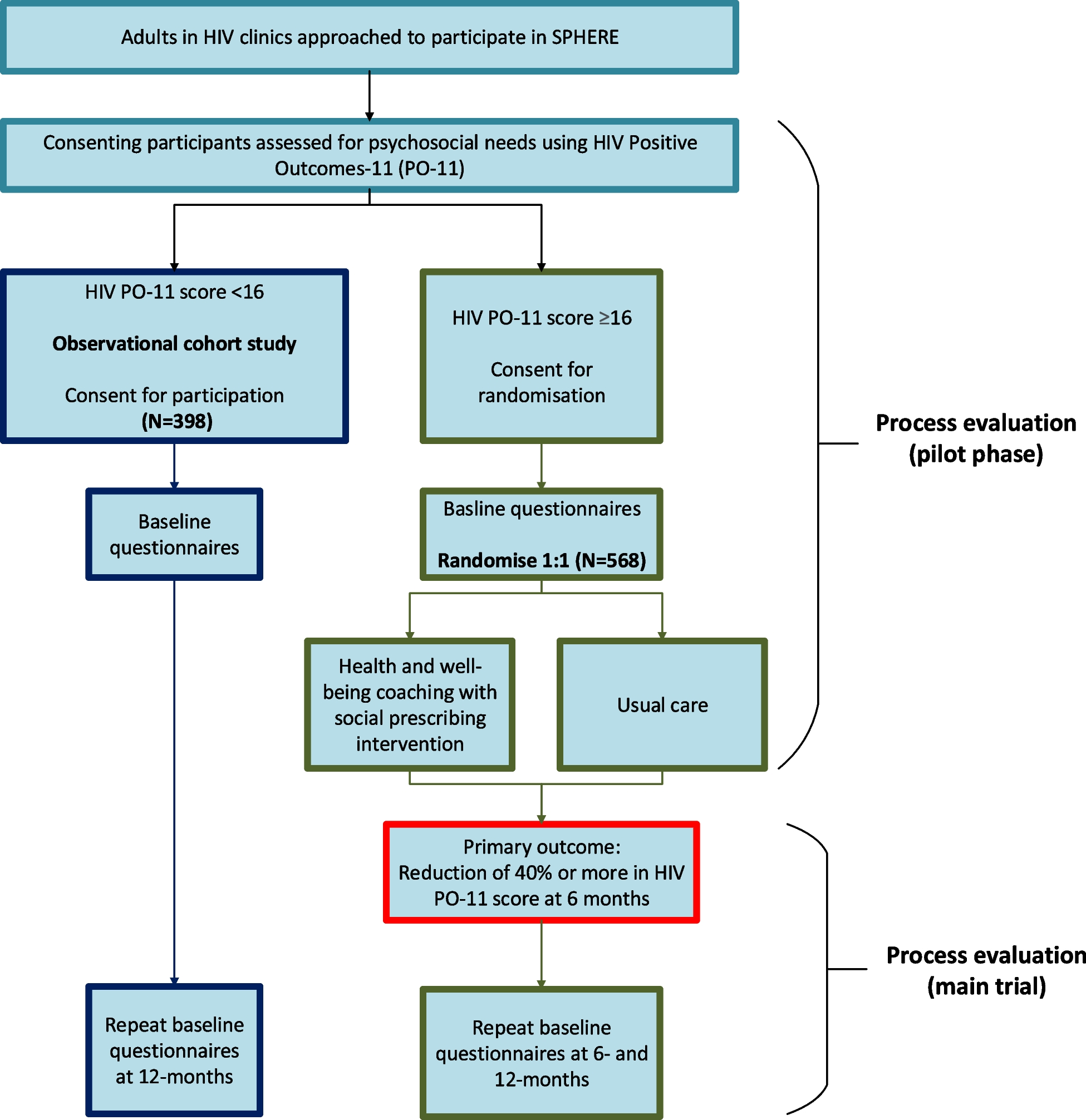
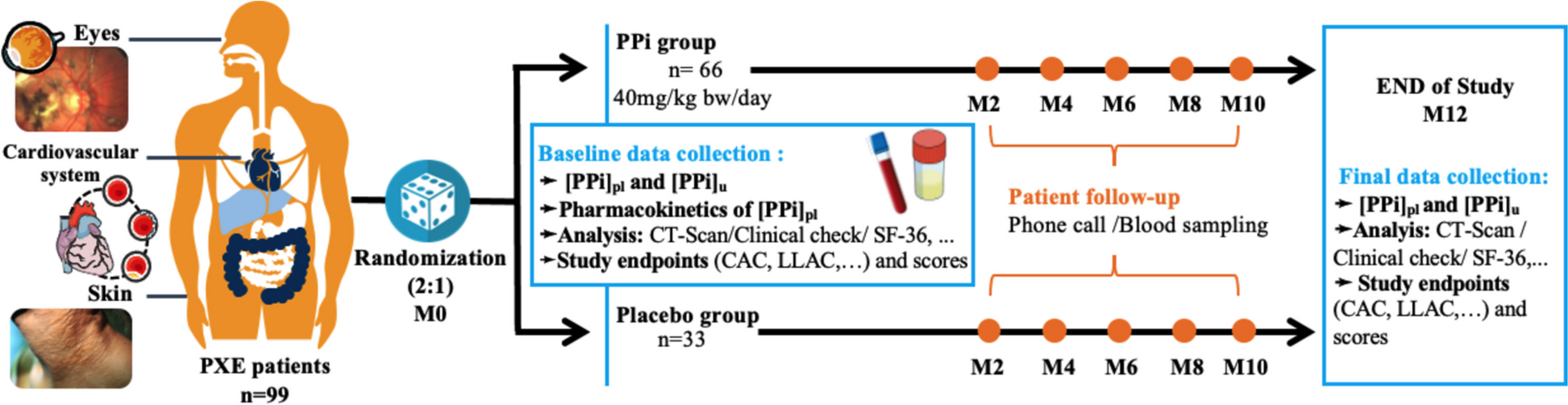



COPERNICOS is a 4-arm facultative designed, randomized controlled, multicenter, parallel group, non-inferiority intervention study in participants with COPD (GOLD class E and/or FEV1 < 30%). The azithromycin versus placebo comparison is double-blinded, while the inhaled corticosteroid (ICS) versus no ICS comparison is open-label due to practical considerations.
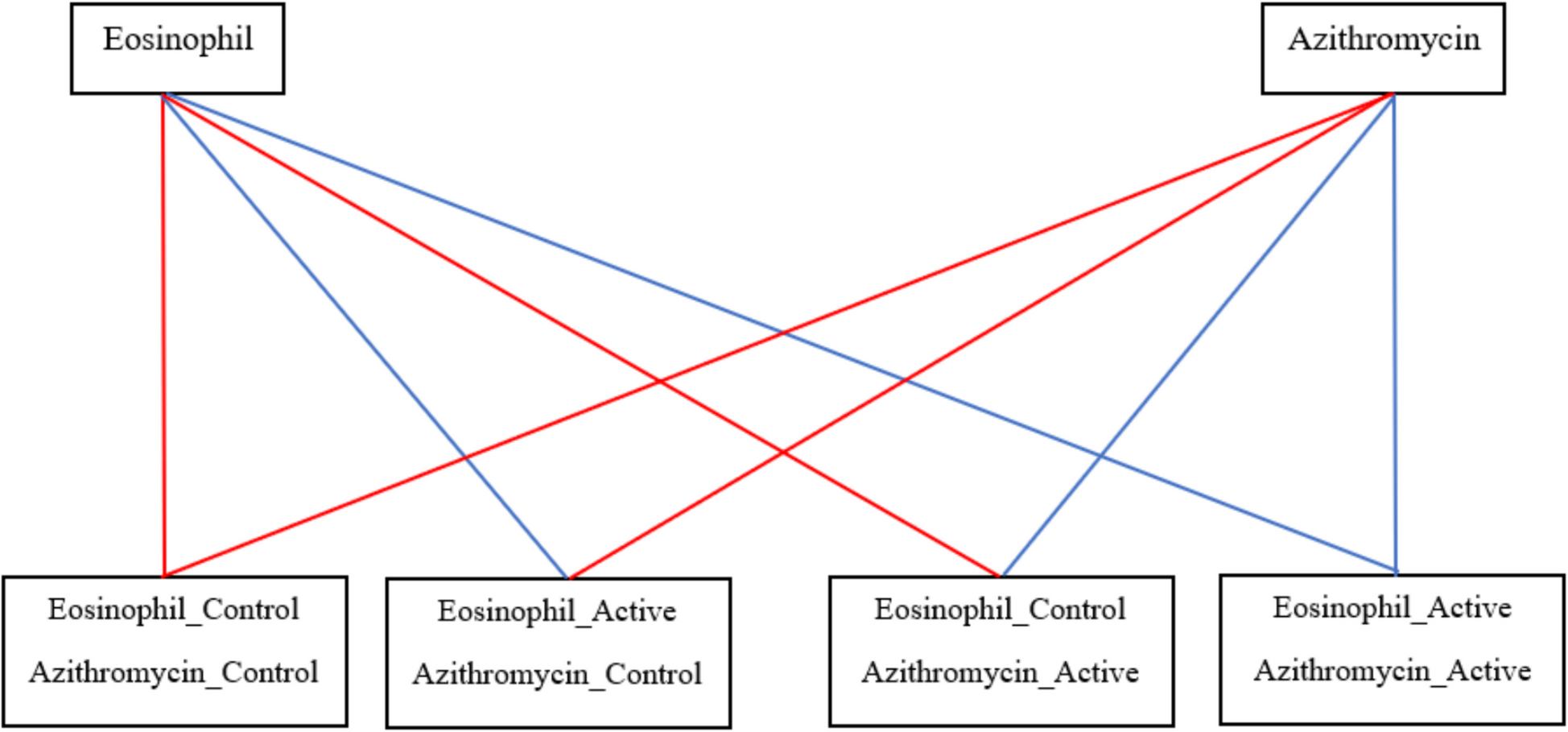
Participants will be randomly allocated to one of the following four treatment groups (Fig. 1):
1.Eosinophil “Control”/Azithromycin “Control” group:
2.Eosinophil “Active”/Azithromycin “Control” group:
3.Eosinophil “Control”/Azithromycin “Active” group:
Azithromycin: 250 mg azithromycin three times weekly.
ICS: The participants are given the usual LAMA/LABA/ICS product in the usual dose throughout the entire project period.
4.Eosinophil “Active”/Azithromycin “Active” group:
Azithromycin: 250 mg Azithromycin three times weekly.
ICS: All participants will receive LABA/LAMA medication. The ICS medication will be switched on/off according to the most recent blood eosinophil count (at inclusion + every 3 months):
i.If blood eosinophil ≥ 0.3 × 109 cells/L, ICS is continued in usual dose for the next 3 months.
ii.If blood eosinophil < 0.3 × 109 cells/L, ICS is discontinued.
Fig. 1
The four treatment groups (Control – Active)
Implementing the intervention will not require alteration to usual care pathways, including the use of any medications. Usual care will continue unchanged in both trial arms. There are no restrictions regarding concomitant care during the trial.
A microbiological and immunological study within the framework of the randomized study aims to investigate the respiratory microbiota and immunological profile in participants from the four randomized groups. A total of 40 participants from each group will participate in the sub-study and will undergo tracheal aspirate collection and nasal swabs at baseline and 12-month follow-up. Collection of tracheal aspirates is a routine procedure at participating centers, and serious complications are very rare. Nasal swab is not associated with any serious complications. Participants’ data and laboratory specimens will be assigned a coded identification number to maintain participant confidentiality. Blood samples for immunological profiling will be collected at baseline and after 12 months of follow-up. We will analyze circulating inflammatory and corticosteroid-responsive biomarkers, including IL-6, IL-8, TNF-α, IFN-γ, eosinophils, and periostin. Additional biomarkers, such as CRP, fibrinogen, and selected transcriptomic/proteomic markers, may be included depending on feasibility. The objective is to explore whether specific immunological signatures correlate with treatment response or adverse events related to ICS.
Modifications to the protocol which may impact on the conduct of the study or potentially affect patient safety, including changes of study objectives, study design, patient population, sample sizes, study procedures, or significant administrative aspects, will require a formal amendment to the protocol. Such amendment will be agreed upon by the COPERNICOS study group and approved by the Clinical Trials Information System (CTIS) prior to implementation.
We used the SPIRIT checklist when writing our report [23].
RecruitmentParticipants for the trial will be recruited through advertisements and announcements on social media, in local newspapers, daily newspapers, and via the Danish Lung Association and its member magazine. Participants can contact trial staff by e-mail or telephone to receive the written participant information. Furthermore, patients at the outpatient clinic at each center will be consecutively screened for eligibility for the trial. If a patient matches the criteria for the study, the patient will be invited to a screening meeting. The oral information session is given by a medical doctor who is duly qualified, including having knowledge of the trial to be able to provide information about it and answer any questions the subject may have. This meeting will take place in an undisturbed office.
Participants will be thoroughly screened for eligibility and informed of the right to an assessor and companion at the first contact during an information meeting with trial staff. A 24-h reflection period will be given to every participant, and informed consent will be obtained upon the signed consent declaration, provided that the participant will participate in the study. Only after receiving oral and written participant information can informed consent be obtained. It is the primary investigator’s responsibility at each center to ensure that study personnel are trained, duly authorized, and competent to obtain informed consent. The consent to collect blood samples, tracheal aspirates, and nasal swabs to research biobank and future-research biobank is a separate part of the informed consent.
At recruitment, health files regarding inclusion and exclusion criteria will be accessed for screening purposes before informed consent is obtained according to the Danish Health Care Act §46 [1]. The following contact to possible participants will be at the next planned visit to the outpatient clinic. During the project, health files will be accessed by investigators for the following information:
Medication
Hospital admissions
If informed consent is obtained, the study sponsor, investigator, monitor, and the Danish Medicines Agency (Lægemiddelstyrelsen) will have access to health files, including electronic files, as part of the trial surveillance, and quality control.
Participants are allocated randomly via REDCap to one of the four arms; prior to randomization, the inclusion and exclusion criteria will have been entered.
The azithromycin/placebo tablets are dispensed either as 500 mg tablets with a score line for splitting (patients are instructed to halve each tablet using the provided pill cutter, or as 250 mg capsules that should be swallowed whole without splitting. Patients will therefore be given very detailed instructions on the provided tablets or capsules and on how to take the medication. The reason for having the medication in two different formulations is that the pharmacy producing the medication, for production reasons, has not been able to supply all the medication in the same formulation. Participants’ compliance to azithromycin/placebo is monitored by both medicine diaries and leftover medicine returned at the 12-month examination. Blood eosinophil count will be monitored every 3 months from inclusion, and participants will receive a phone call from a nurse every 3 months to ensure that participants have taken ICS according to the physician’s prescription and plan the next 3 months’ treatment according to protocol.
Inclusion criteriaCOPD (verified by a specialist in respiratory medicine + spirometry).
GOLD class E anytime within the last 2 years (corresponding to 2 ≥ AECOPD and/or ≥ 1 AECOPD leading to hospitalization during a 12 month period within the last 2 years) and/or FEV1 < 30%.
Treatment for last 4 weeks including LAMA, LABA and ICS.
Informed consent.
Exclusion criteriaKnown asthma.
Male < 40 years.
Female < 40 years, if non-menopausal (had menstruation within the last 12 months) conditioned by a negative urine HCG test
Severe mental illness which considerably complicates co-operation.
Language problems that considerably complicate co-operation.
Current treatment with systemic corticosteroids corresponding to > 5 mg prednisolone per day.
Systemic antibiotic treatment (if to participate in microbiota sub-study) or systemic corticosteroid treatment within 14 days (also prophylactic azithromycin).
Contra-indication to treat with azithromycin (as listed by the producer).
Non-bacterialexacerbation per investigator judgement in the last 3 months.
*Non-bacterial exacerbation per investigator judgement may be guided by
Exacerbation with blood eosinophil > 0.3 × 109 cells
Exacerbation with CRP < 100 mg/L
Exacerbation with chest x-ray without sign of pneumonia
ExaminationsThe following clinical tests will be performed during the project cf. Fig. 2.
Fig. 2
An overview of examinations that each participant will undergo
Blood tests: Electrolyte parameters (sodium, potassium, albumin, creatinine, hemoglobin), liver parameters (conjugated bilirubin, ALT, alkalic phosphatase, INR, LDH), infection parameters (CRP, white blood cell differential count, thrombocytes), D-dimer, and HbA1c.
Questionnaires: Standardized questionnaires are used. CAT and MRC dyspnea scale are short and simple tests that provide an understanding of the severity and impact of COPD on the participant’s daily life.
Data collectionData processing, statistical analysis, and publication of the data material will be performed by a PhD student (coordinating investigator) together with health professionals (primary and secondary investigators) from the participating pulmonary medical outpatient clinics. The randomized trial started with the inclusion of participants on 05 JUL 2021, and the last participant is expected to be included with follow-up ending by 31 NOV 2025. Data collection ceases on 31 NOV 2025, and the project is scheduled to run until 31 DEC 2026. A nurse and a doctor will be responsible for collecting baseline data, performing lung function measurement, weight, and height at baseline and the 12-month follow-up, as well as performing blood sampling and questionnaires at three-, six-, and nine-month follow-ups, according to Fig. 2. Data will be obtained via the electronic health record. Medical decisions are made by doctors only. Proper handling, storage, and delivery of medication and the primary daily project management are handled by the coordinating investigator. Recruitment, medical examinations, and distribution and accounting of medication will be assisted by primary investigators at each center.
The data collected will be treated confidentially and only by personnel associated with the project. This includes demographic data, health status, current illnesses, medications, medicinal side effects, hospital admissions, and results from various examinations during the trial. Data will be reported in Electronic Case Report Forms (eCRF) specific to each participant. The data is encrypted, stored in online servers, and protected by the Data Protection Authority through various security precautions. The physical copies of the CRF are kept in locked archives in the involved departments for 5 years.
Data in eCRF will be handled by the investigator at each center and in accordance with the Law for Data Protection and the Danish Law for Privacy Regulation.
The coordinating center, based at Copenhagen Respiratory Research, is responsible for the overall organization of the trial, including data management, monitoring, regulatory compliance, and communication across sites. It provides the central infrastructure and coordination needed to implement the protocol effectively.
Any important modifications to the protocol (e.g., changes to eligibility criteria, outcomes, or analysis plans) will first be communicated to the trial sponsor and funder. Upon approval, the principal investigator (PI) will inform all participating trial centers. A copy of the revised protocol will be sent to each site for inclusion in the Investigator Site File. Any deviations from the protocol will be fully documented using a breach report form. Furthermore, any changes to the protocol will be promptly updated in the relevant clinical trial registry.
The day-to-day conduct of the trial will be coordinated by the Project Management Group (PMG), consisting of trial investigators, clinical leads, research staff, trial managers, and data managers from the coordinating center at Copenhagen Respiratory Research. The PMG is responsible for overseeing recruitment, data collection, monitoring adherence to the protocol, and resolving operational issues. The group will meet monthly throughout the recruitment and follow-up periods.
The trial will also be overseen by a Trial Steering Committee, comprising independent clinical experts, trial methodologists, and a chairperson, who will provide external oversight and ensure that the trial is conducted in accordance with the highest scientific and ethical standards. The Trial Steering Committee will meet biannually to review trial progress, protocol adherence, data quality, and safety concerns. There is currently no formal Stakeholder and Public Involvement Group; however, patient representatives will be engaged in the interpretation and dissemination of results.
Research biobankThe purpose of establishing a research biobank is to investigate the frequency of ICS-induced side effects and the changes in respiratory microbiota in different treatment groups. This biobank will clarify the hypotheses and provide biological materials for future research (Providing that future projects can obtain a separate approval from the Danish National Committee on Health Research Ethics).
The research biobank will store blood samples, tracheal aspirate samples, and nasal swabs. Tracheal aspirate and nasal swab samples are taken from 40 participants from each treatment arm, in total 160 participants. Approximately 1–5 ml of tracheal aspirate will be collected from each participant. Blood samples are taken from each participant at inclusion and at follow-up every 3 months during the 12-month study period. Approximately 210 ml of blood will be collected during the entire study period corresponding to a maximum of 36 ml per blood draw. All samples are stored in a freezer at –80 °C. The freezers are kept in a locked room at each of the participating pulmonary medicine departments.
All samples are pseudonymized and kept for 15 years in accordance with present legislation and data protection laws. Establishment of this research biobank ends 31 AUG 2027. Following the end of this study and the research biobank, all surplus biological material will be transferred to a future-research biobank. These samples will also be pseudonymized and kept for 15 years. Permission to store biological material from participants in the research biobank and future-research biobank is a part of the informed consent to participation in the project.
Comments (0)