Primers, plasmids, bacterial strains, and cell lines
A detailed list of the primers utilized in the present study can be found in Table S1. A collection of plasmids essential for our experiments was procured from Hunan Fenghui Biotechnology Co., Ltd., China. These included pEGFP-N1, pCAG-Cre: GFP, pUC19, pACYC177, pcDNA™3.1/myc-His A, pSPAX2, pMD2.G, pLVX-IRES-Puro, pLVX-IRES-mCherry, and pLVX-IRES-BFP. Notably, the plasmid pUC57-intein, which contains the sequences of intein, was synthesized by Beijing Tsingke Biotech Co., Ltd., China. Additionally, we used the following Addgene plasmids as generous gifts from Pawel Pelczar [21]: pCAG-NLS-HA-Dre (Addgene plasmid # 51272), pCAG-roxSTOProx-ZsGreen (Addgene plasmid # 51274), and pCAG-loxPSTOPloxP-ZsGreen (Addgene plasmid # 51269). The E. coli strain DH5α was utilized as a general cloning host for molecular cloning and β-galactosidase activity assays. The human embryonic kidney cell line 293T was used to evaluate the recombinase activity of Dre and Cre, as well as for virus packaging. 4T1 mouse mammary carcinoma cells were used to assess the recombinase activity of Dre and Cre.
Culture and growth conditions
The Escherichia coli strain DH5α was cultured in Luria broth (LB) media and on LB agar plates at 37 °C. For plasmid maintenance, the growth medium was supplemented with ampicillin (100 µg/ml) and/or kanamycin (25 µg/ml) as specified in the experimental procedures. In addition, 293T cells were cultured in high-glucose DMEM supplemented with 10% fetal bovine serum (FBS) and antibiotics (penicillin and streptomycin). 4T1 cells were maintained in RPMI-1640 medium containing 10% FBS and antibiotics (penicillin and streptomycin). Both cell lines were incubated at 37 °C with 5% CO2.
DNA constructs
pLVX-IRES-Puro-roxEGFP(rox) vector construction
The EGFP cassette, flanked by two opposing rox sequences, was PCR-amplified from pEGFP-N1 via primers (rox)-F and (rox)-R. The products were subsequently cloned and inserted into pLVX-IRES-Puro at the XhoI and BamHI sites via the Seamless Cloning Kit (Beyotime Biotechnology, China).
pcDNA™3.1/myc-His a vectors for Dre and Cre genes and variants
We constructed pcDNA™3.1/myc-His A vectors containing Dre and Cre genes and their variants via PCR amplification from pCAG-NLS-HA-Dre and pCAG-Cre plasmids, respectively. The primers used are listed below, and the products were subsequently cloned and inserted into pcDNA™ 3.1/myc-His A at the EcoRI and XbaI sites.
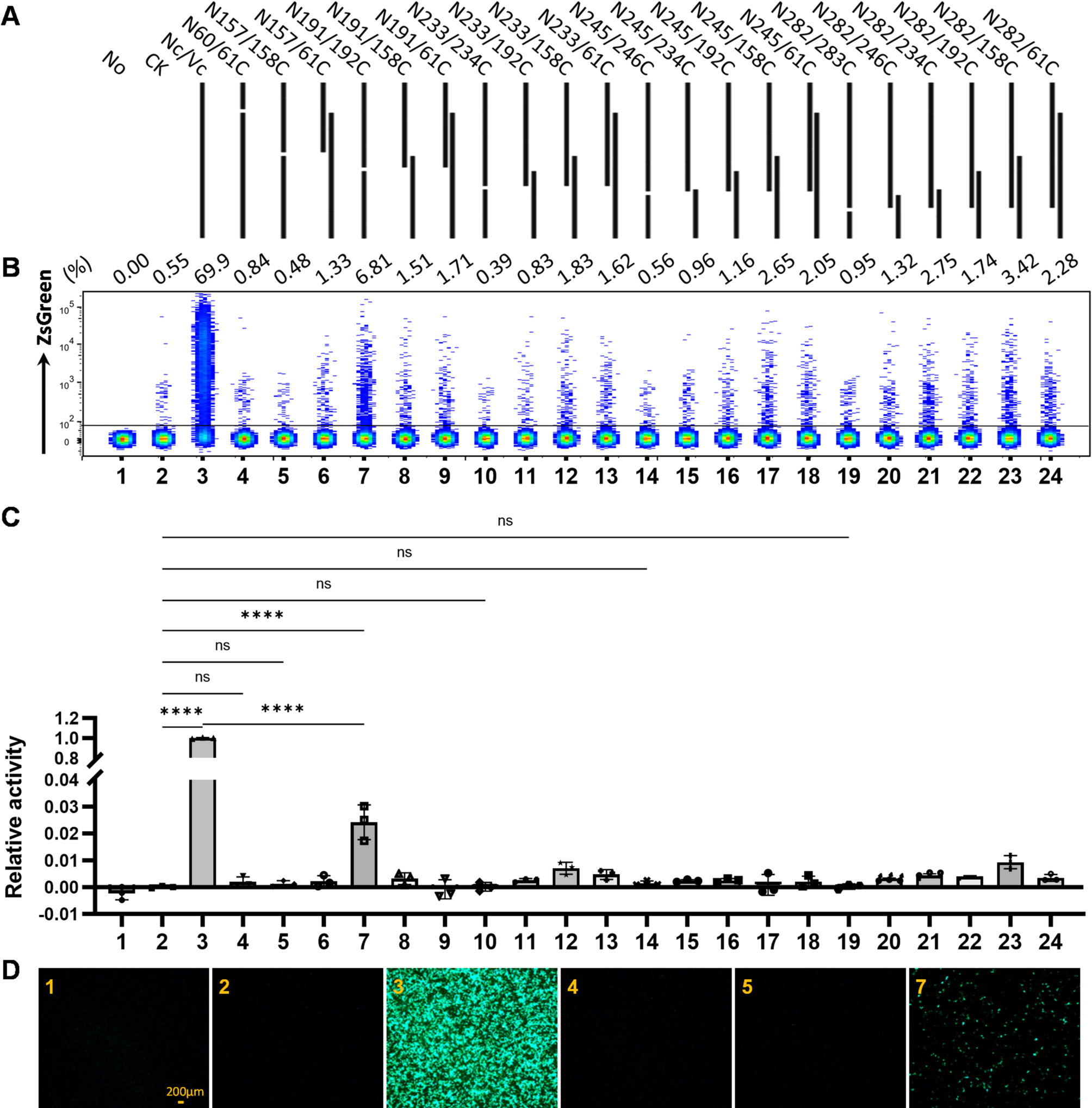
Dre Gene Variants: Dre-F and Dre-R (Dre gene, Nc), Dre-F and Dre60-R (N60), Dre-F and Dre157-R (N157), Dre-F and Dre191-R (N191), Dre-F and Dre233-R (N233), Dre-F and Dre245-R (N245), Dre-F and Dre282-R (N282), 61Dre-F and Dre-R (61C), 158Dre-F and Dre-R (158C), 192Dre-F and Dre-R (192C), 234Dre-F and Dre-R (234C), 246Dre-F and Dre-R (246C), 283Dre-F and Dre-R (283C).
Cre Gene Variants: Cre-F and Cre-R (Cre gene, Nc), Cre-F and Cre59-R (N59), Cre-F and Cre190-R (N190), 60Cre-F and Cre-R (60C), 191Cre-F and Cre-R (191C).
The pLVX-IRES vectors with the Dre gene and variants
pLVX-IRES-mCherry and pLVX-IRES-BFP vectors containing the Dre gene and its derivatives were constructed via PCR amplification from pCAG-NLS-HA-Dre. The primers used were as follows, and the products were subsequently cloned and inserted into the respective vectors at the EcoRI and XbaI sites.
pLVX-IRES-mCherry: Dre-F and Dre-R (Dre gene, NC), NLSDre-F and Dre-R (NNC), NLSDre-F and DreNLS-R (NNCN), NLSDre-F and Dre60-R (NN60), NLSDre-F and Dre191-R (NN191).
pLVX-IRES-BFP: 61Dre-F and Dre-R (61C), 61Dre-F and DreNLS-R (61CN), 192Dre-F and Dre-R (192C), 192Dre-F and DreNLS-R (192CN).
pLVX-IRES vectors with Dre variants and intein
We constructed pLVX-IRES-mCherry and pLVX-IRES-BFP vectors with Dre variants fused to intein via seamless cloning. The primers used for the Dre and intein fragments are detailed below, and the products were subsequently cloned and inserted into the respective vectors at the EcoRI and XbaI sites.
NN60nI: S-NLSDre-F and Nintein-Dre60-R (Dre), Dre60-Nintein-F and S-Nintein-R (intein).
NN191nI: S-NLSDre-F and Nintein-Dre191-R (Dre), Dre191-Nintein-F and S-Nintein-R (intein).
Ic61C: Cintein-61Dre-F and S-Dre-R (Dre), S-Cintein-F and 61Dre-Cintein-R (intein).
Ic192C: Cintein-192Dre-F and S-Dre-R (Dre), S-Cintein-F and 192Dre-Cintein-R (intein).
Ic61CN: Cintein-61Dre-F and S-NLSDre-R (Dre), S-Cintein-F and 61Dre-Cintein-R (intein).
Ic192CN: Cintein-192Dre-F and S-NLSDre-R (Dre), S-Cintein-F and 192Dre-Cintein-R (intein).
pUC19 vectors with Dre variants
pUC19 vectors incorporating Dre variants were generated via seamless cloning. The primers used for amplification were as follows, and the products were cloned and inserted into pUC19 at the BamHI and EcoRI sites.
Dre gene (nc): p19-Dre-F and p19-Dre-R.
N60-61C: p19-Dre-F and p19-Dre60-R (N-terminal), p19-61Dre-F and p19-Dre-R (C-terminal).
N191-192C: p19-Dre-F and p19-Dre191-R (N-terminal), p19-192Dre-F and p19-Dre-R (C-terminal).
pACYC177-roxLacZα(rox) vector construction
The LacZα cassette, flanked by two opposing rox sequences, was PCR-amplified from pUC19 via (rox)LacZ-F and (rox)LacZ-R. The products were subsequently cloned and inserted into pACYC177 at the PstI site via the Seamless Cloning Kit (Beyotime Biotechnology, China).
Cell line generation
Lentivirus for EGFP expression was generated by co-transfecting 293T cells with the plasmids pLVX-IRES-Puro-roxEGFP(rox), psPAX, and pMD2.G at a ratio of 4:3:1. Stable cell lines expressing the reverse EGFP gene were established in 293T and 4T1 cells via lentiviral-mediated transduction. Following transduction, cells were selected for stable integration through puromycin (6 µg/ml) selection for three weeks to ensure long-term expression of the transduced gene.
Flow cytometry analysis of EGFP/ZsGreen-expressing 293T or 4T1 cells
Single-cell suspensions of transfected 293T and 4T1 cells were prepared and washed once with PBS. The samples were analyzed using a BD LSRFortessa™ flow cytometer. Data were analyzed using FlowJo (BD, v10.8.1). The percentage of EGFP/ZsGreen-expressing cells was compared by concatenating the samples of live transfected cells.
Recombinase activity assay for Dre, Cre and their derivatives
Plasmids encoding Dre, Cre, and their derivatives, at a concentration of 0.5 µg/24well, were transfected into 293T or 4T1 cells utilizing the jetPRIME® reagent (Polyplus). Following transfection, the proportion of cells expressing fluorescent proteins was assessed by flow cytometry at designated time intervals. The fluorescence intensity within transfected 293T cells was quantified using the Spark multimode microplate reader (Tecan). Additionally, representative images were acquired through an Olympus CKX53 inverted microscope equipped with a DP22 camera and CellSens software.
Immunoblot analysis
Cells were lysed in RIPA buffer (P0013B; Beyotime Biotechnology, China) to extract total protein. The lysates were subsequently homogenized and centrifuged at 12,000 g at 4 °C for 15 min to remove cellular debris. The supernatants were collected for further analysis. The samples were analyzed by western blotting using the standard procedures [31]. The targeted protein was detected by immunoblot with the following primary antibodies: anti-Dre (NBP3-11868; Novus Biologicals, CO, USA) and anti-β-Tubulin (TA-10; ZSGB-Bio, China), which served as a loading control. ECL western blotting detection reagents (PK1003; Proteintech, China) were used to visualize the protein bands, and the images were captured using a Tanon 5500 Chemiluminescent Imaging System.
β-galactosidase activity assay
The β-galactosidase activity of the Escherichia coli strain DH5α harboring the plasmids described in the text was determined according to the protocol of the β-Galactosidase Assay Kit (Beyotime Biotechnology, China), with slight modifications. Active bacterial proteins were extracted from 0.2 mL of bacterial cultures via BeyoLytic™ Bacterial Active Protein Extraction Reagent (Beyotime Biotechnology, China). The samples were incubated with the β-galactosidase detection reagent for a period of 4 h. The samples were subsequently centrifuged at 10,000 rpm for 10 min in a microcentrifuge to pellet the cell debris. The absorbance of the supernatant at 420 nm (A420) was then measured to quantify the β-galactosidase activity, with a blank control prepared from the parental strain without a plasmid used to account for background absorbance. For β-galactosidase activity assays on LB plates, X-Gal was added.








Comments (0)