The collection and extraction of the experimental plant
The leaf of Peristrophe bivalvis was gathered from its natural habitat on privately owned land in the South-South region of Nigeria, particularly within the Ikono local government area of Akwa-Ibom State in the month of June 2018, following verbal permission from the landowner. The verification and identification process was conducted by Mr. Okon Udo Etefia, a botanist in the Department of Pharmacognosy and Herbal Medicine at the University of Uyo, Akwa-Ibom State, and the specimen was assigned the batch number ID-ALUKO/UUHO43/IKONO.
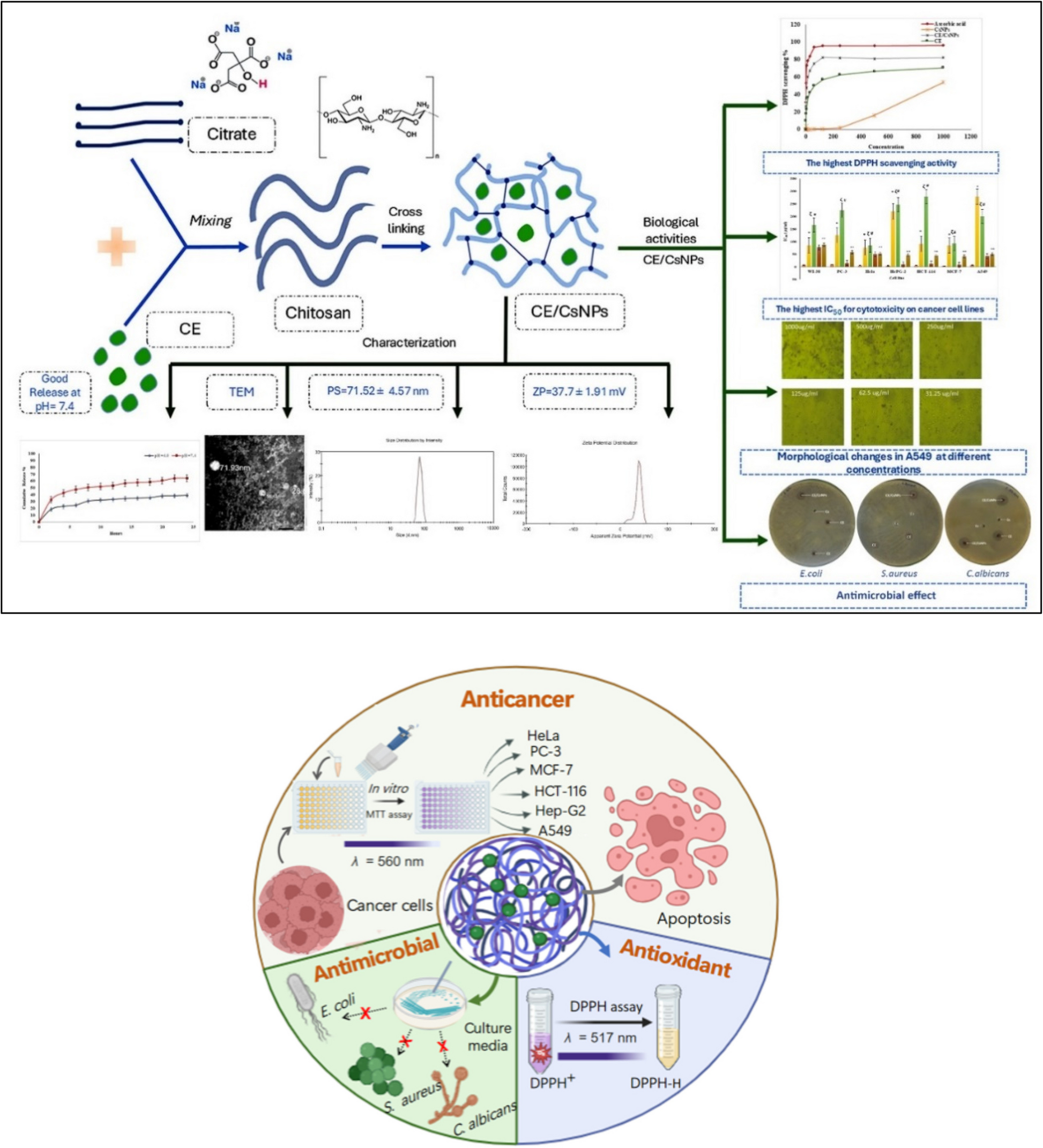
The aqueous extract of Peristrophe bivalvis leaf was obtained as follows: The leaves were washed with tap water and distilled water, then air-dried at 25 °C. The dried leaves were pulverized into powder, and then 1000 g of this powder was marinated in 10 L of distilled water for three days with consistent stirring. After three days, the mixture was filtered using muslin cloth and Whatman filter paper. The filtrate was concentrated using a rotary evaporator set at 35 °C, followed by freeze-drying. The final yield obtained was 130 g at a percentage yield of 13%, which was appropriately labeled and stored in the refrigerator at 4 °C until required for use. The stock solution was prepared daily. The aqueous extract of Peristrophe bivalvis leaf at the dose of 200 mg/kg body weight was suspended in distilled water before being orally administered to the animals.
Induction of hypertension
Hypertension was induced by inhibiting the production of nitric oxide (NO) with a non-selective nitric oxide synthase (NOS) inhibitor (Nw-nitro-L-arginine methyl ester [L-NAME]). This was administered orally to the rats (60 mg/kg/day) for eight weeks. The blood pressure of the animals was measured while conscious using the tail-cuff occlusion technique at the beginning, at the eighth week, and at the end of the experiment. L-NAME was obtained from DYC Group Ltd., Hangzhou City, China. L-NAME was dissolved in distilled water and administered to the animals via oral gavage using an oral cannula.
Research animal
Thirty (30) male Wistar rats weighing between 150 and 170 g (10–12 weeks old) were used. They were procured from the University of Ibadan Central Animal House and housed in standard cages placed in a secure and conducive environment in the College of Medicine Animal House. The animals had free access to standard rat chow and water, and a 12-hour light and 12-hour dark cycle at 25 ± 3 °C was maintained throughout the duration of the study. A two-week acclimatization period was observed before the study commenced. Additionally, one week was also used to train the animals to familiarize them with the blood pressure measurement setup.
The research procedures were approved and assigned a reference number- UI-ACUREC/2017/058 by the Animal Ethics Committee, University of Ibadan, Nigeria. Throughout the entire period of the study, the animals were cared for and handled according to the Animal Ethics Committee’s guidelines. This study adheres to internationally accepted standards for animal research, following the 3Rs principle. The ARRIVE guidelines were employed for reporting experiments involving live animals, promoting ethical research practices.
Experimental design
The present study used thirty male Wistar rats, which were randomly divided into six groups with five (5) animals per group:
Group 1: Control group (C).
Group 2: Hypertensive sacrificed in week 8 (HS8).
Group 3: Untreated hypertensive group (H).
Group 4: Hypertensive recovery group (ReH).
Group 5: standard drug treated hypertensive group (RaH).
Group 6: Aqueous extract of Peristrophe bivalvis leaf (APBL) treated hypertensive group (APBLH).
The dosages for L-NAME (60 mg/kg b.w.), ramipril (10 mg/kg b.w.), and APB (200 mg/kg b.w.) used in this study were all selected based on previous studies: studies by Martinez-Fierro et al. [24], Zhu et al. [25], and KocYildirim et al. [26] for L-NAME; Adeyinka et al. [27], Dugbartey et al. [28], and Al-Kadi et al. [29] for ramipril. The dosage for APB was determined based on our previous study [23]. Additionally, the qualitative phytochemical analysis, in vitro antioxidant activity, and gas chromatography-mass spectrometry assessment of APB have been reported in our earlier study [23]. The number of animals per group was based on the guidelines of the Animal Ethics Committee at the University of Ibadan, Nigeria.
The control group was orally gavaged with 10 mL/kg b.w. of distilled water, while groups 2 and 6 were orally gavaged with 60 mg/kg b.w. of L-NAME daily for eight weeks to induce hypertension. After eight weeks, animals in group 2 were sacrificed. Groups 3, 5, and 6 continued to receive 60 mg/kg b.w. of L-NAME with 10 mL/kg b.w. of distilled water, 10 mg/kg b.w. of ramipril, and 200 mg/kg b.w. of APB, respectively, for an additional five weeks. Group 4 received only 10 mL/kg b.w. of distilled water during the five weeks of treatment. The induction of hypertension and treatment comprised a total of 13 weeks of study.
Blood pressure assessment
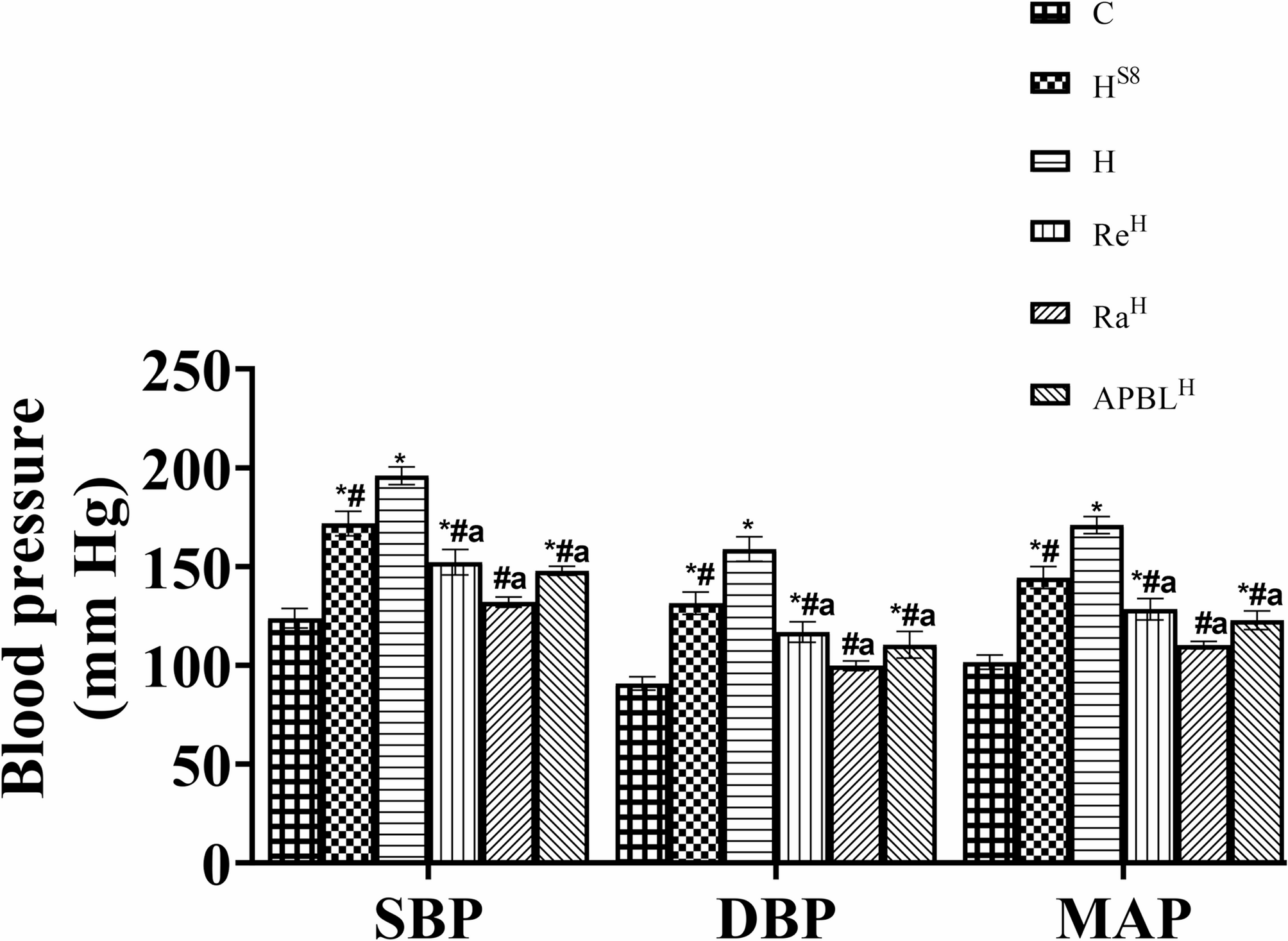
Blood pressure was assessed by a non-invasive method using the tail-cuff occlusion technique. The setup used was the CODA, Kent Scientific, USA as described by Lee et al. [30]. Prior to the experiment, the animals underwent training for a week to become familiar with the blood pressure setup. Furthermore, the blood pressure measurement was taken at the same time of day in each measuring week, and during every assessment, the room temperature was maintained within the range of 32 to 35 °C. Briefly, the rat was placed in a restraining device while conscious, which was positioned on a heated (35 °C) platform; the occlusion cuffs (volume pressure and occlusion cuffs) were fastened around the animal’s tail. Before the commencement of the measurement, the animal observed a 15-minute acclimation period. The assessment consisted of five acclimatization cycles succeeded by fifteen real cycles. The average of all recorded measurements (at least three) recognized by the CODA system, excluding the acclimatization cycles and values considered outliers for each rat, was taken as the blood pressure.
Biochemical and ELISA assessment
Blood sample was collected by cardiac puncture from anesthetized (0.1 ml/100 g b.w. ketamine/xylazine IP.) rat into a plain bottle, then centrifuged, and the serum was collected for biochemical and ELISA estimations. Following the collection of blood samples, the rats were euthanized using an excessive dose of ketamine/xylazine, after which the heart, kidneys, and thoracic aorta were harvested from the euthanized animals. The tissues were immersed in 10% formaldehyde for histological and immunohistochemical studies. For biochemical and ELISA tests, the tissues were initially washed in cold phosphate-buffered saline (pH 7.4) and then dried with filter paper. Subsequently, they were homogenized with a Teflon homogenizer in phosphate-buffered saline (pH 7.4), and the resulting homogenate was centrifuged using cold centrifugation at 10,000 x g for 10 min at 4 °C.
Nitric oxide concentration was measured indirectly by estimating the level of nitrite using the spectrophotometric technique described by Ghosh et al. [31]. A reagent (Griess reagent) was added to the sample and incubated for 30 minutes. The absorbance of the mixture was measured at a wavelength of 548 nm. To evaluate lipid peroxidation, the concentration of malondialdehyde was quantified by measuring thiobarbituric acid reactive substances levels, following the methodology outlined by Özdemir et al. [32]. Malondialdehyde reacts with thiobarbituric acid to produce a pink mixture, whose intensity is measured at 535 nm. The spectrophotometric method was used to measure the concentrations of glutathione (GSH), catalase (CAT), and superoxide dismutase (SOD). SOD concentration was measured with an SOD assay kit from R&D Systems, Inc., Minneapolis, USA, and the CAT level was determined with a CAT assay kit from Sigma, St. Louis, USA. The concentration of glutathione was assessed by quantifying the levels of reduced glutathione [33].Reduced glutathione contains non-protein sulfhydryl groups that react with Ellman’s reagent (5’, 5’-dithiobis-(2-nitrobenzoic acid)) to form a stable yellow complex. This complex’s absorbance is measured at 412 nm wavelength.
Creatinine and urea levels were measured using creatinine and urea assay kits (Fortress Diagnostics Limited, Antrim, UK), respectively, and all procedures were conducted according to the manufacturer’s instructions. The liver enzymes- aspartate transaminase (AST), alanine transaminase (ALT), and alkaline phosphatase (ALP) levels were evaluated using Randox diagnostic assay kits, as described by Hasan et al. [34]. The manufacturer’s guidelines were meticulously followed. For AST evaluation, the serum sample was mixed with a solution containing L-aspartate, phosphate buffer, and L-oxoglutarateand incubated for 30 min at37°C. Then, a solution containing 4, dinitrophenyl hydrazine and sodium hydroxide (NaOH) was added and incubated for 20 min, after which NaOH was added and left for 5 min. The absorbance was measured at 540 nm. ALT level was determined by mixing the serum sample with a solution containing DL-alanine, α-oxoglutarate, and phosphate buffer and incubated for 30 min. Then, a solution containing 2, 4 dinitrophenyl hydrazine and NaOH was added and incubated for 20 min, after which NaOH was added and left for 5 min. The complex’s absorbance is measured at 412 nm wavelength. ALP level was determined by colorimetric method by mixing the serum sample with a working solution provided by the manufacturer, and the intensity was measured at 405 nm.
The enzyme linked immunosorbent assay (ELISA) technique was used to determine the levels of endothelial nitric oxide synthase (eNOS), endothelin-1 (Elabscience Biotechnology Inc., Corporation, USA), atrial natriuretic peptide (ANP), and angiotensin II (ANG II) (MyBiosource Incorporated, San Diego, USA). The manufacturer’s guidelines were meticulously followed. Similar method is applied universally to all ELISA tests. It utilizes the principle of competition. The protocol previously described by Tabatabaei and Ahmed [35] was used in this study. The absorbance of all the assays was measured at a wavelength of 450 nm.
Examination of heart histology
The histological examination of cardiac tissue was conducted utilizing hematoxylin and eosin (H&E) staining techniques as previously described [36]. The tissues were sliced, mounted on slides, and stained with H&E. Photomicrographs were captured at 400× magnification. The severity of morphological alterations was graded using a semi-quantitative scoring system. The scale 0–3 was used to score inflammation, thrombosis, and fibrosis: 0 = normal without inflammation, thrombosis, or fibrosis; 1 = mild inflammation, thrombosis, or fibrosis; 2 = moderate inflammation, thrombosis, or fibrosis; 3 = severe inflammation, thrombosis, or fibrosis.
Determination of vWF, KIM-1, and nitrotyrosine expression
The expressions of vWF in the aorta and KIM-1 in the kidney were assessed using immunohistochemical technique. Every immunohistochemistry staining follows similar procedures. Immunohistochemistry is a method for identifying antigens in a tissue section, employing the principle of antibodies binding to their specific antigens within the tissues. This antibody-antigen interaction is made visible by 3, 3’-diaminobenzidine (DAB), which produces a brown-colored reaction [37].
Briefly, for vWF and KIM-1, the tissue blocks were fixed in Bouin’s fluid, and a 5 μm section was cut from each tissue block using a microtome. These sections were mounted on positively charged slides for immunohistochemical staining. The slides were placed in 0.01 M citrate buffer (pH 6.0) for 10 min at 120 °C, after which they were placed in methanol containing 0.3% hydrogen peroxide for 15 min. They were then washed with phosphate-buffered saline, and blocking buffer was pipetted onto the slides and incubated for 30 min at 25 °C. A solution containing 1 µg/mL primary monoclonal and polyclonal antibodies, as well as 2 µg/mL phosphate-buffered saline, was pipetted onto the slides and left for 30 min. This was followed by three consecutive washings with phosphate-buffered saline. Then, the secondary antibody was pipetted onto the slides and incubated for 30 min, after which they were washed three times with wash buffer. Following this, metal-enhanced 3,3’-diaminobenzidine was pipetted onto the slides and left for 10 min. The slides were washed with wash buffer twice and counterstained with hematoxylin stain. Finally, the photomicrographs were captured, and the expressions of vWF in the aorta, KIM-1 in the kidney, and nitrotyrosine in the aorta, heart, and kidney were quantified using ImageJ software (version 1.49, National Institutes of Health, Bethesda, MD, USA.
Statistical analysis
The results of this study were presented as mean ± SD (standard deviation). They were analyzed using one-way and two-way analysis of variance (ANOVA). Tukey’s post-hoc test was employed for comparison between groups. The GraphPad Prism 8.01 software (GraphPad Software, Inc., USA) was utilized for the aforementioned statistical analyses. Statistical significance was accepted at p < 0.05.




Comments (0)