
Remember me



Renal glomerular podocytes, which line the outer surface of glomerular capillary tufts, constitute the last filtration interface that restrains protein leakage during ultrafiltration. Damage to these cells is a defining clinical sign of chronic kidney disease, including hypertensive nephropathy [1].
Over recent decades, mechanistic studies of podocyte injury have predominantly emphasized non-hemodynamic drivers such as hormonal or metabolic stressors. By contrast, advances in mechanobiology—particularly the identification of mechanosensitive ion channels—have refocused attention on hemodynamic stress as a direct molecular determinant of podocyte injury [2, 3].
Among mechanosensitive ion channels, Piezo1 has emerged as a key regulator of podocyte responses to mechanical stress [4]. While several studies have examined the functional role of Piezo1 in glomerular disease models, their findings have been inconsistent. In non-hypertensive settings, podocyte-specific Piezo1 knockout mice showed either protective effects in lupus [5] and diabetic nephropathy [6], or aggravated injury in Adriamycin nephropathy [7], underscoring the context-dependent nature of Piezo1 signaling. However, Ogino et al. demonstrated that Piezo1 expression increases in podocytes exposed to hypertensive stress, where mechanical stretch–induced activation of Piezo1 triggers injury-related transcriptional and cytoskeletal responses [8]. While these findings implicated Piezo1 activation in hypertensive podocyte injury, they did not determine whether the channel itself is required to preserve glomerular integrity under mechanical stress.
To address this unresolved question, the study by Mikami et al. in this issue [9] generated podocyte-specific Piezo1 knockout (p-Piezo1 KO) mice and systematically assessed the consequences of Piezo1 loss under both basal and hypertensive conditions. Two complementary models were applied to capture distinct temporal aspects of hypertensive stress: a short-term angiotensin II infusion with high-salt intake (AII + HS) to induce acute glomerular pressure elevation, and a chronic uninephrectomy/aldosterone/high-salt (UNx/Ald/HS) regimen to model sustained mineralocorticoid-driven injury. This dual approach allowed the authors to examine both short- and long-term podocyte responses to mechanical overload.
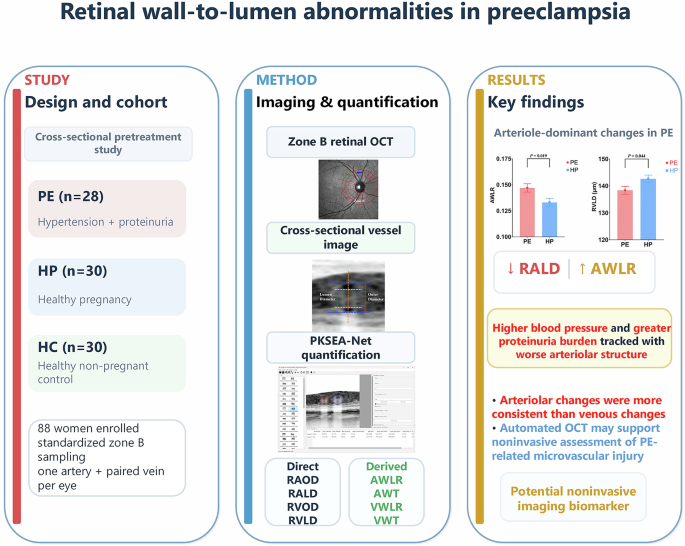
Under physiological conditions, p-Piezo1 KO mice appeared normal and maintained stable renal function, indicating that Piezo1 is dispensable for glomerular homeostasis at rest. In contrast, hypertensive loading unmasked a clear vulnerability: knockout mice developed marked albuminuria, diffuse foot-process effacement, and glomerulosclerosis compared with wild-type littermates. Importantly, systemic blood pressure elevation was comparable between genotypes, excluding confounding by hemodynamic load. Ultrastructural analysis revealed extensive foot-process fusion and slit-diaphragm detachment, together reflecting a loss of cytoskeletal resilience under stress. Immunostaining showed reduced synaptopodin and nephrin expression, underscoring structural destabilization of the filtration barrier. Collectively, these findings position Piezo1 as a key regulator of cytoskeletal integrity that enables podocytes to endure hypertensive mechanical stress.
The authors further deepened the mechanistic understanding through multi-layered molecular and pharmacologic analyses. Glomerular RNA-seq under basal conditions, in the absence of hypertensive stress, identified 35 differentially expressed protein-coding genes enriched in cytoskeletal and adhesion pathways, including upregulation of Rhpn1 (Rhophilin1, Rho GTPase binding protein 1), Fgfbp1 (Fibroblast growth factor binding protein 1), and Gadd45a (Growth arrest and DNA damage–inducible 45 alpha)—all linked to actin regulation—and downregulation of Dlk2 (Delta-like non-canonical Notch ligand 2). These transcriptional alterations observed at baseline suggest that Piezo1 contributes to the maintenance of cytoskeletal homeostasis even in the absence of external mechanical stress. Rho-family GTPases, particularly RhoA and Rac1, are known to orchestrate actin remodeling and podocyte structural stability under mechanical cues [10]. Pharmacologic inhibition of Rho-kinase with fasudil, but not Rac1 inhibition, partially ameliorated albuminuria and structural damage, implicating excessive RhoA-driven actomyosin contraction in Piezo1-deficient podocytes. Together, these results integrate morphologic, transcriptomic, and pharmacologic insights to define Piezo1 as a mechanoprotective hub maintaining balance between RhoA- and Rac1-dependent cytoskeletal dynamics in podocytes.
How can this protective phenotype be reconciled with previous reports of Piezo1-mediated injury? A coherent view is that Piezo1 signaling operates within an optimal range: chronic overactivation under severe mechanical or metabolic stress can drive sustained Ca²⁺ influx with cytoskeletal collapse and oxidative stress, whereas complete loss removes adaptive mechano-buffering, shifting the system toward cytoskeletal stiffening. This bidirectional model explains how deviations in either direction—excessive or insufficient Piezo1 activity—can compromise podocyte integrity.
From the perspective of Rho GTPases, the present data indicate that Piezo1 deficiency biases signaling toward RhoA–Rho-kinase–dependent actomyosin tension, reducing the Rac1-associated membrane plasticity that normally permits adaptive remodeling under load. Several lines of evidence converge on this interpretation: (i) basal glomerular transcriptomics revealed upregulation of Rhpn1 and enrichment of cytoskeletal/adhesion pathways, consistent with RhoA scaffolding; (ii) Rho-kinase inhibition partially rescued albuminuria and structural injury, whereas Rac1 inhibition was ineffective; and (iii) ultrastructural analyses showed diffuse foot-process fusion and slit-diaphragm detachment, features compatible with excess contractile tone. These findings align with prior mechanobiology work in podocytes showing that mechanical cues engage Rho-family GTPases to coordinate actin architecture and adhesion dynamics [2, 3].
Excessive Rac1 activity has been implicated in podocyte injury through multiple stress pathways. Previous work linked Rac1 activation to mineralocorticoid receptor (MR)–dependent injury and proteinuria [11,12,–13], while another showed that Piezo1-mediated Rac1 activation promotes NFAT nuclear translocation and TRPC6 upregulation, enhancing calcium influx and cytoskeletal stress in diabetic kidney disease [6]. These findings indicate that Rac1 overactivation can disrupt cytoskeletal homeostasis through both MR- and calcium-dependent mechanisms, whereas RhoA dominance under Piezo1 loss leads to excessive contractility and impaired adaptive remodeling.
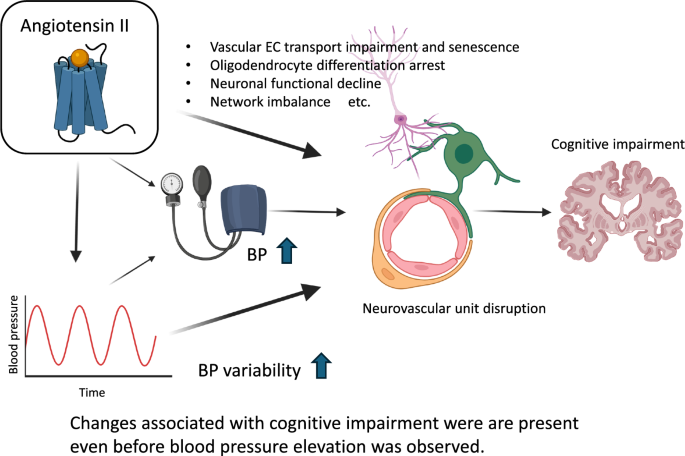
This bidirectional vulnerability underscores the concept that Piezo1 functions as a rheostat maintaining cytoskeletal balance between RhoA- and Rac1-mediated programs (Fig. 1). Partial rescue by Rho-kinase inhibition supports the notion that fine-tuning, rather than complete suppression, of mechano-signaling is therapeutically beneficial.
Fig. 1
Schematic illustration of the proposed mechanism linking Piezo1 to cytoskeletal regulation in podocytes under hypertensive stress. This figure illustrates how mechanical stress activates Piezo1 to maintain balanced signaling between the RhoA and Rac1 pathways, thereby stabilizing the actin cytoskeleton. In Piezo1 knockout mice, loss of this mechanosensory control shifts the balance toward RhoA–Rho kinase (ROCK)–mediated actomyosin contraction, resulting in cytoskeletal stiffening and podocyte injury under hypertensive conditions. Pharmacologic inhibition of Rho kinase—a downstream effector of RhoA that promotes contractility—with fasudil partially ameliorates this phenotype
Beyond pathological contexts, these findings also raise fundamental questions about the physiological role of Piezo1-mediated mechanosensation in glomerular homeostasis. Under normal hemodynamic load, podocytes must continuously remodel their actin cytoskeleton to accommodate cyclic stretch and filtration pressure. Piezo1 may serve as a molecular transducer that senses these subtle mechanical oscillations and calibrates the balance between RhoA-driven stability and Rac1-dependent flexibility. Loss of this mechanoadaptive feedback, as seen in the knockout models, could predispose podocytes to injury even before overt hypertension develops.
These insights also carry translational implications. Given that conventional hemodynamic therapies only indirectly influence podocyte stress, strategies that selectively modulate the Piezo1–RhoA–Rac1 axis could provide organ-specific protection without systemic hypotension. The partial efficacy of Rho-kinase inhibition observed here hints that targeted attenuation of contractile signaling, combined with preservation of mechanosensory adaptability, may represent a feasible therapeutic paradigm. Defining the molecular determinants that distinguish adaptive from maladaptive Piezo1 activation will be an important next step toward precision modulation.
The strength of this study lies in its integrative design. By combining a cell-specific knockout strategy with two independent hypertensive models, detailed ultrastructural imaging, transcriptomic profiling, and pharmacologic interventions, the authors advanced previous work on podocyte mechanotransduction from in vitro stretch assays to in vivo functional genomics. Identifying Rhpn1 as a plausible effector gene adds molecular depth, linking Piezo1 loss to altered RhoA-signal scaffolding.
Nonetheless, several questions remain open for exploration. Bulk glomerular RNA‑seq, while informative, may dilute cell‑specific transcriptional signatures; single‑cell or spatial transcriptomics could clarify whether Rhpn1 upregulation is podocyte‑intrinsic. Biochemical assays of active RhoA (e.g., G-LISA or pull-down) would confirm pathway engagement, and rescue experiments with Piezo1 re‑expression might establish causality. From a translational perspective, validating and expanding the observed correlation between PIEZO1 and RHPN1 in human kidney disease datasets—such as hypertensive nephrosclerosis or diabetic kidney disease—could further strengthen the clinical relevance of these mechanistic pathways.
In conclusion, the study by Mikami et al. [9] advances our understanding of how podocytes integrate mechanical cues into structural resilience. Piezo1 emerges not as a simple on–off switch but as a context dependent integrator that tunes cytoskeletal tone through RhoA/Rac1 equilibrium. These findings extend the conceptual view of Piezo1 from a potential driver of stretch induced injury to a necessary component of adaptive mechanotransduction under hypertension. Future therapies may thus seek to recalibrate, rather than silence, mechanosensitive signaling in the glomerulus.
Comments (0)