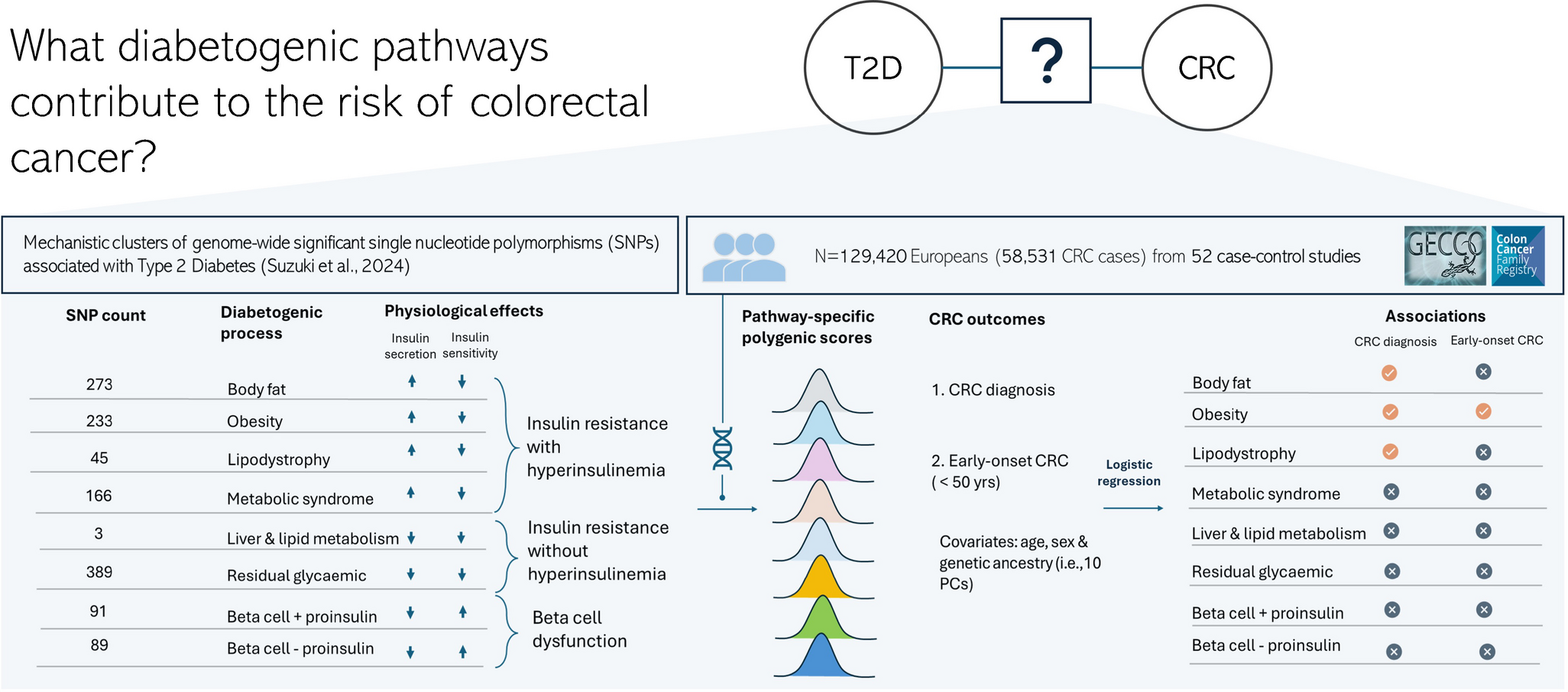
In this large-scale study of over 129,000 individuals from the GECCO and CCFR consortia, we demonstrate that diabetogenic processes characterised by insulin resistance-linked hyperinsulinaemia are associated with increased odds of CRC. In contrast, we found no evidence of significant associations for pathways related to relative insulin secretion insufficiency or insulin resistance without hyperinsulinaemia. In addition, we showed that the PPS for obesity-related hyperinsulinaemic insulin resistance was associated with early-onset CRC, particularly among individuals with elevated BMI. Taken together, these findings advance the mechanistic understanding of the relationship between type 2 diabetes and CRC, and highlight the potential for targeted risk stratification and prevention.
Our study builds on extensive epidemiological evidence demonstrating an association between type 2 diabetes and increased CRC risk [3, 5, 6, 15, 16, 24] by dissecting the contribution of specific diabetogenic processes. Previous observational studies have yielded conflicting results, with some reporting stronger associations with shorter type 2 diabetes duration, implicating early metabolic disturbances such as hyperinsulinaemia in CRC [5, 6], while others have found stronger links with longer diabetes duration, possibly reflecting cumulative metabolic damage [16]. These divergent findings probably reflect the clinical and molecular heterogeneity of type 2 diabetes, with varying degrees of residual insulin secretion and insulin sensitivity across individuals and over time [25, 26]. A recent Mendelian randomisation study supported a causal effect of hyperinsulinaemia on increased CRC risk [10]. These results are aligned with those of previous experimental studies that demonstrated that hyperinsulinaemia can enhance colorectal epithelial proliferation and tumorigenesis through adipose tissue-released factors and impaired insulin and IGF signalling [11,12,13]. Chronic low-grade inflammation, which frequently accompanies insulin resistance-linked hyperinsulinaemia, may represent an additional mechanism linking diabetogenic pathways to CRC risk [27], and should be considered in future mechanistic and epidemiological investigations. Our findings expand on these observations by providing suggestive evidence that insulin resistance-linked hyperinsulinaemia processes, particularly those mediated through adipose tissue mechanisms, are associated with increased odds of CRC, including early-onset disease.
A major strength of this study is the use of PPSs to investigate the diabetogenic mechanisms underlying the associations between type 2 diabetes and CRC. These scores were constructed using genetic variants that have established associations with specific metabolic phenotypes that have been validated through tissue-specific epigenomic and gene expression data, offering biologically meaningful representations of type 2 diabetes pathophysiology. This integrative approach yields functionally grounded representations of the underlying metabolic pathways implicated in type 2 diabetes pathophysiology. Our findings showing that insulin resistance-linked-hyperinsulinaemia mediated through obesity, lipodystrophy and body fat mechanisms is associated with increased odds of CRC represent an advance over previous studies showing that hyperinsulinaemia is causally associated with CRC. Of particular interest, we observed that certain well-characterised genes that are implicated in hyperinsulinaemia, such as INSR, IRS1 and IGF1R, are exclusively represented within clusters defined by insulin resistance-linked hyperinsulinaemia, which supports the biological plausibility of our findings, and provides a mechanistic context for understanding the observed associations.
A novel and clinically important aspect of this study is the link to early-onset CRC, a form of the disease with increasing incidence and poorly understood aetiology [6, 28]. Emerging evidence indicates that early-onset CRC may present unique mutational, epigenetic and metabolic features. A previous study from our consortium that included 6176 patients with early-onset CRC and 65,829 control participants identified new early-onset CRC susceptibility genes related to insulin signalling and immune/infection-related pathways [29]. Others have reported that early-onset CRC may exhibit distinct molecular features, including microsatellite instability, a CpG island methylator phenotype, and mutations in TP53 and PTEN, while BRAF mutations are less common than in older-onset CRC [30]. These molecular features probably interact with environmental and metabolic factors, manifesting in susceptible individuals. In our study, only the obesity-related hyperinsulinaemic insulin resistance PPS was associated with early-onset CRC. This association was independent of behavioural factors such as smoking and alcohol intake but was significantly modified by BMI: individuals with obesity and high genetic susceptibility had a 75% higher odds of early-onset CRC compared with those at lowest genetic risk. No evidence of associations was observed among normal-weight individuals. These findings highlight the importance of gene–BMI interactions in early-onset CRC, and suggest that genetically determined metabolic dysfunction may play a key role in early tumorigenesis in this increasingly common CRC subtype.
Although the effect sizes observed in our study were modest, with ORs ranging from 1.1 to 1.3 when comparing individuals in the top vs bottom deciles of the PPSs, these estimates are broadly consistent with the 1.2–1.5-fold increased CRC risk reported in large observational studies of type 2 diabetes [3]. Reporting effect size based on extremes of genetic risk is supported by evidence from a recent population-based prostate cancer screening programme. In that study, targeted screening of participants in the highest 10% of genetic risk identified clinically actionable prostate cancer in 55.1% (n=103) of cases, and cancer would not have been detected in 74 of these participants (71.8%) according to current prostate cancer diagnosis pathways [31]. Our study highlights the potential utility of PPSs to help identify individuals at elevated CRC risk, particularly those who do not meet current age-based screening guidelines. Younger adults with obesity and those with metabolically adverse genetic profiles may represent an under-recognised high-risk population that could benefit from targeted screening, as reflected in the recent international management guidelines initiative for early-onset CRC [32]. Although our work was not designed to inform clinical risk prediction or screening strategies, these insights contribute to a deeper understanding of the shared pathophysiology between diabetogenic processes and CRC, and highlight opportunities for eventual mechanistically informed screening and prevention strategies.
Several limitations merit consideration. First, replication in independent studies is needed to confirm the generalisability of our findings. Second, the study population was primarily of European ancestry, which may limit applicability to other populations. However, the latest multi-ancestry GWAS of CRC has shown limited population-specific heterogeneity [21]. Furthermore, the pathway-specific PPSs used in this study have shown consistent associations with cardiovascular outcomes across ancestries, suggesting that the underlying biological mechanisms may be broadly conserved [18]. Third, while the case–cohort design employed here is well suited for aetiological inference, it may limit direct applicability to the broader population. Future prospective observational studies are necessary to better assess the predictive capability of these PPSs in CRC risk. Fourth, hyperinsulinaemia itself can act as a causal driver of adiposity, suggesting a bi-directional relationship between insulin secretion and body fat. This complexity has implications for interpreting our findings, as genetic determinants that promote hyperinsulinaemia may increase CRC risk both directly, through mitogenic signalling pathways, and indirectly, through their effect on adiposity. Our stratified analyses by BMI provide some insight, showing effect maximisation in individuals with high BMI, consistent with the presence of both direct and indirect effects. However, future studies incorporating longitudinal metabolic and imaging data are essential to disentangle these pathways and clarify the relative contributions of hyperinsulinaemia and adiposity to CRC risk. Finally, we lacked data on potentially important confounders or effect modifiers, such as socioeconomic status, dietary habits, healthcare access and CRC screening behaviours (e.g. colonoscopy use), which could influence the observed associations [5]. Future studies should aim to incorporate these variables to further refine risk estimates.
In conclusion, our findings suggest that diabetogenic processes characterised by insulin resistance-linked hyperinsulinaemia are associated with increased odds of CRC, including early-onset disease. In contrast, no such associations were observed for relative insulin secretion insufficiency or insulin resistance without hyperinsulinaemia, underscoring the specificity of the implicated diabetogenic processes in CRC pathogenesis. Furthermore, we observed a synergistic interaction between genetic susceptibility and elevated BMI, underscoring the importance of gene–environment interactions in early-onset CRC. While replication in ancestrally diverse populations and further evaluation of clinical utility are needed, these results have important public health and clinical implications. They support a use of a more mechanistically informed approach to CRC prevention and early detection, particularly in individuals with obesity or adverse genetic risk profiles.

Comments (0)