
Remember me


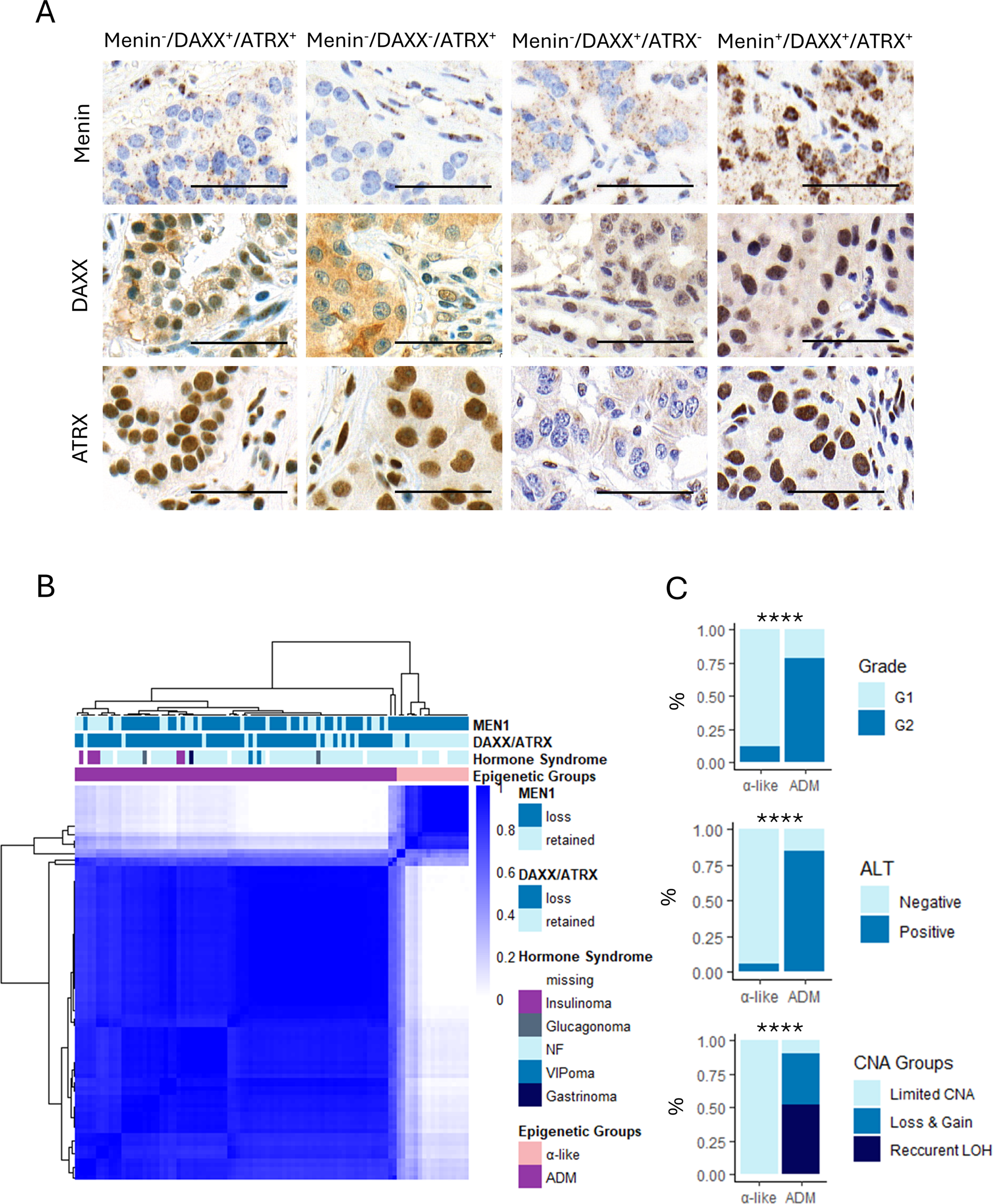
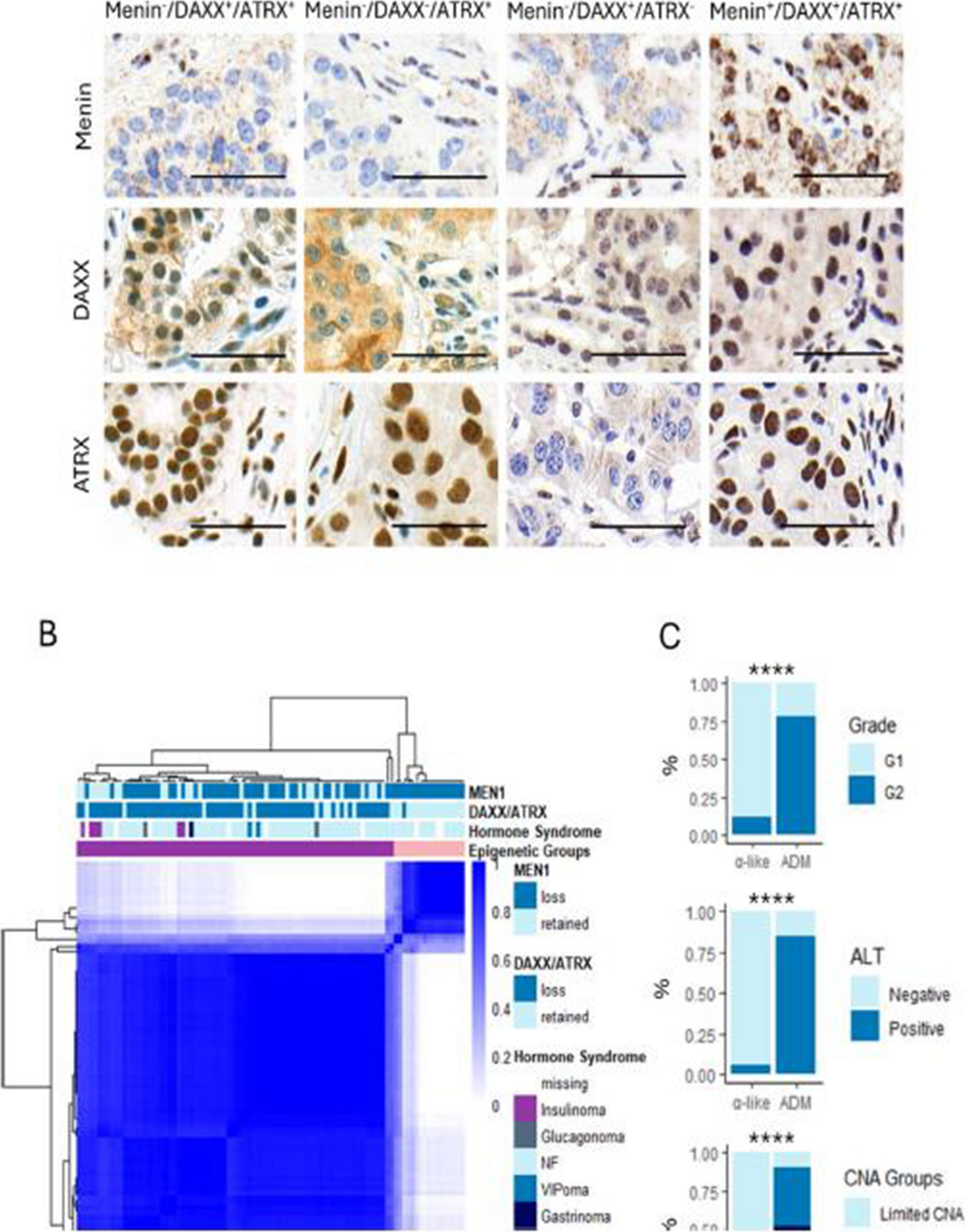
To dissect the heterogeneity of the ADM PanNET subtype, we performed DNA methylation analysis of samples with loss of nuclear expression of at least one of the α-cell–associated proteins: Menin, DAXX, or ATRX (n = 93, Fig. 1A).
Fig. 1
PanNETs with loss of DAXX/ATRX and Menin can be divided into 2 groups according to DNA methylation. A Representative immunohistochemistry of loss (−) and retention (+) of Menin, DAXX, and ATRX. Each column represents one sample; each row corresponds to immunohistochemistry staining of the reported protein. Scale bars in the right bottom represent 50 μm. B Consensus clustering of the 93 PanNETs with loss of DAXX/ATRX and/or Menin loss according to the top 2000 most variably methylated probes (selected by MAD) shows segregation of α-like and ADM PanNETs. Consensus cluster correlation is indicated in the heatmap with darker colors representing stronger correlation. Each column represents one sample. Menin and DAXX/ATRX status, hormone syndrome, and epigenetic subtypes are indicated according to the reported colors. C ADM PanNETs are strongly associated with higher grade, alternative lengthening of telomeres (ALT), and copy number alteration (CNA) (Pearson’s Chi-squared test, ****p-value < 0.0001). LOH = loss of heterozygosity
To recapitulate the α-like and ADM PanNET epigenetic subtypes, we performed consensus clustering using the 2000 most variably methylated probes [2, 20]. When employing two clusters, DNA methylation profiles clearly distinguish subtypes which are strongly associated with loss of DAXX/ATRX expression, in accordance with previous findings [2]. Therefore, the cluster enriched in tumors with loss of Menin, but not DAXX/ATRX, was labelled α-like, while the cluster enriched in samples with loss of Menin and DAXX/ATRX was referred to as ADM (Fig. 1B). Importantly, ADM PanNETs were associated with higher grade (Pearson’s Chi-squared test, p.value = 7.0 × 10−6), activation of alternative lengthening of telomeres (ALT, Pearson’s Chi-squared test, p.value = 4.0 × 10−8), and chromosomal instability (Pearson’s Chi-squared test, p.value = 2.3 × 10−8), indicating more aggressive phenotype (Fig. 1C). The vast majority of included samples were non-functioning (n = 72), followed by insulinomas (n = 6), VIPomas (n = 2), and one gastrinoma. Out of the six insulinomas, four were positive for ARX and negative for PDX1, one was negative for ARX and positive for PDX1, and for one sample the immunohistochemistry was not available.
Hypomethylation of Telomeric and Peri-Centromeric Regions is Associated with Increased Chromosomal Instability in ADM PanNETsTo investigate which specific epigenetic changes occur between α-like and ADM PanNETs, we performed differential methylation analysis, which revealed 6316 CpG probes (DMPs, p.adj < 0.001, |Δβ| >0.2, Supplementary Table 2).
We interrogated the location of these probes in the islet-specific genomic feature atlas defined by Thurner et al. [39]. Over-representation analysis revealed enrichment in enhancers and lowly-methylated regions, suggesting that the changes between α-like and ADM tumors affect mainly non-coding regions (Fig. 2A).
Fig. 2
Characteristic DNA methylation differences between the three NF-PanNET subgroups. A Graphical representation of DMP distribution across chromatin states based on over-representation analysis (one-tailed Fisher’s exact test). Odds ratio > 1 (above dotted line) indicates region enriched in the DMPs. Adjusted p-values for each region are -log10 transformed and indicated according to the color key. B and C Median methylation signal of subtelomeric (B) or peri-centromeric (C) regions are significantly lower in ADM PanNETs (t-test, *p-value < 0.05, **p-value < 0.01). Each dot represents one sample. Presence of alternative lengthening of telomeres (ALT) or copy number alteration (CNA) is indicated according to color legend. NA designates samples for which ALT or CNA status was not available. LOH- loss of heterozygosity
Hypermethylation of heterochromatin, especially at subtelomeric and pericentromeric regions, is crucial for maintenance of chromosomal stability [40]. To investigate the association between epigenetic changes, ALT and copy number alteration (CNA) in PanNETs we assessed the median methylation levels of sub-telomeric and peri-centromeric regions. Both regions showed higher median methylation in the α-like compared to the ADM PanNETs (Fig. 2B and C), linking the loss of methylation in structural regions with ALT activation and genomic instability. Methylation levels of specific chromosomes on sub-telomeric and peri-centromeric regions are depicted in Supplementary Fig. 2A and Supplementary Fig. 2B, respectively. Notably, chromosomes 3,5,7,9,11,14,17,19, and 20 are hypomethylated in subtelomeric regions, while chromosomes 1,2,6,7,11,12,13,14,17, and 22 are hypomethylated in peri-centromeric regions.
Progression from α-like to ADMEpigenetic and genetic evidence suggests that ADM tumors could develop from α-like tumors upon DAXX/ATRX loss of function [2].To assess this possibility, we conducted consensus clustering including an additional set of nine matching metastases (Supplementary Table 1, Supplementary Fig. 1A and 1B). One sample pair was treated with neoadjuvant chemotherapy (streptotozin-doxorubicin) before resection of both primary tumor and metastasis (Subject 123). All other pairs were treatment naïve. Consensus clustering used the same probes that distinguished α-like and ADM PanNETs also targeting two clusters. Interestingly, one sample pair segregated between α-like and ADM tumors (subject 168, Fig. 3A). Notably, the primary tumor clustering with α-lineage PanNETs showed ATRX retention, while the matching metastasis clustered with the ADM tumors and was ATRX negative (Fig. 3A and B). Furthermore, ALT was negative in the primary tumor, but positive in the metastasis (Fig. 3D and E, respectively). These results provide evidence for progression from α-like to ADM NF-PanNET in one patient.
Fig. 3
Alpha-line metastatic NF-PanNET shows epigenetic progression. A Consensus clustering of 93 primary PanNETs and 9 matching metastases according to the 2000 most variable probes from Fig. 1B. Consensus clustering dendogram was obtained for k = 2. The yellow to blue color scale indicates the DNA methylation value (β value, 0 = unmethylated, 1 = methylated) for each probe (in rows). Each column represents one sample. Matching patient samples and tissue type are indicated according to the color legend. Subject 168 (in green) shows progression from α-like primary tumor into ADM metastasis. B Telo-FISH shows activation of alternative lengthening of telomeres in metastasis (white arrows), but not in primary tumor. Immunohistochemistry for ATRX is preserved in primary tumor and lost in metastasis. Both primary tumor and metastasis show Menin loss and DAXX retention
Transcriptomic Analysis Distinguishes 4 Subtypes of ADM PanNETNext, we performed gene expression analysis for a subgroup of 36 samples for which we had RNA-seq data available in addition to the DNA-methylation data (Supplementary Fig. 1, Supplementary Table 1).
Consensus clustering of these samples using the 2000 most variably expressed genes (selected by MAD) did not recapitulate the epigenetic α-like trait. The α-like PanNETs did not form a separate cluster (Supplementary Fig. 3A), but were intermixed in the ADM PanNETs, suggesting that transcriptomic profiling alone is insufficient to distinguish between the two entities. Since the primary focus of this study was to investigate heterogeneity within ADM PanNETs, we excluded the five α-like PanNETs from further analysis, resulting in a final set of 31 ADM samples.
Consensus clustering on the 2000 most variably expressed genes (selected by MAD) confirmed transcriptomic heterogeneity of ADM PanNETs (Fig. 4A). Good cluster stability could be obtained with 4 clusters (ADM-1 to ADM-4). Higher resolutions further increased stability but also resulted in the creation of additional small clusters (Supplementary Fig. 3B and C). Of note, the cluster 4 (ADM-4) forms very early (k = 2) and remains stable until k = 10, underscoring its specific transcriptomic profile (Supplementary Fig. 3C). However, due to the small sample size (n = 2) it had to be excluded from further analysis. Our analysis did not reveal any differences in risk of metastasis formation, relapse, and perineural or vascular space invasion between the four ADM groups (Supplementary Fig. 3D-G). However, the ADM-3 PanNETs were significantly enriched in tumors smaller than 2.5 cm (Fig. 4A, chi-squared test with simulated p value, p value = 0.023, median = 2.4 cm), and the ADM-1 seem to have higher Ki67 albeit not significant likely due to the small sample size (Fig. 4A, Supplementary Fig. 3H). While the majority of included PanNETs are non-functioning, we have also three insulinomas (two in ADM-2, one in ADM-3), two VIPomas (one clustering with ADM-4, one with ADM-1), and one glucagonoma in ADM-1. Of note, two insulinomas and the glucagonoma were ARX positive and PDX1 negative, as assessed by immunohistochemistry. The immunohistochemistry was not available for the VIPomas and one insulinoma.
Fig. 4
ADM PanNETs are transcriptionally heterogeneous. A Consensus clustering of ADM PanNETs based on 2000 most variably expressed genes (selected by MAD). Each column represents a sample. Darker colors in the heatmap represent stronger cluster correlation. Hormone syndrome and transcriptomic subtypes are indicated according to the color key (NF- non-functioning). B Heatmap of scaled mean expression (in rows) of transcription factors linked with α-, β-, γ-, δ-, and ε-cell per consensus cluster (in columns). Transcriptomic subtypes are indicated by color. Blue and red color in the heatmap represents low and high expression, respectively. C-E Gene set enrichment analysis based on DEGs identified for each group. Top 10 up-regulated hallmark pathways (indicated on the left) are depicted. Adjusted p-value colored by the color key. F MLP1 gene signature score (y axis) per group (x axis). Boxplots are colored according to the transcriptomic groups
Investigation of islet-cell specific transcription factors (TFs) defined by Muraro et al. revealed differences among the groups [41]. The Kruskal–Wallis test confirmed significant differences in the expression of transcription factors associated with α- (adjusted p.value = 0.049), β- (p.value = 0.033), and δ- (p = 0.0241) cells across the ADM groups. Specifically, ADM-2 exhibited the highest expression of transcription factors linked to α- and β-cells, ADM-1 was enriched for δ-cell transcription factors, and ADM-3 showed elevated expression of those associated with γ- and ε-cells. (Fig. 4B).
In order to characterize the specific ADM subtypes, we performed differential gene expression analysis between each group and the rest of the cohort. We identified similar number of differentially expressed genes (DEGs, |log2FC|>1, adjusted p-value < 0.05) for ADM-1 (870, Supplementary Table 3) and ADM-2 (899, Supplementary Table 4). ADM- 3 rendered the lowest number of DEGs (498, Supplementary Table 5).
Interrogation of Hallmark pathways enriched in individual clusters revealed characteristic metabolic changes in ADM-1, notably up-regulation of hypoxia and glycolysis (Fig. 4C). Therefore, we refer to ADM-1 cluster as ADM hypoxic. Other up-regulated pathways were involved in cell cycle (E2F targets, G2M checkpoint), epithelial to mesenchymal transition (EMT), mTOR, KRAS or TNFA signaling, complement and coagulation (Fig. 4C). ADM- 2 showed different metabolic changes (oxidative phosphorylation, fatty acid metabolism, and adipogenesis). Of note, this cluster comprises three functioning tumors, including two insulinomas, which could explain the enrichment in pancreatic β-cells and protein secretion pathways (Fig. 4D) and increased expression of β-cell TFs (Fig. 4B). Other pathways encompass bile acid metabolism, peroxisome, and mitotic spindle (Fig. 4D). Since this cluster did not present any specific characteristics, we named it ADM NST (no special type). ADM-3 showed enrichment mainly in inflammation and immune-related pathways (allograft rejection, inflammatory and interferon γ response, and IL2 and IL6 signaling) (Fig. 4E). Therefore, we labelled it ADM immunosuppressive.
Importantly, Young and colleagues previously characterized MLP1 subtype with strong hypoxic and immunosuppressive transcriptional phenotype [15]. In accordance, ADM hypoxic and ADM immunosuppressive show higher MLP1 transcriptomic signature compared to the ADM NST (Fig. 4F).
ADM Hypoxic Signalling is Epigenetically DrivenNext, we sought to understand whether the identified transcriptomic changes could be driven by DNA methylation of promoters. Therefore, we investigated the correlation between methylation of CpG sites and corresponding differentially expressed genes (adjusted p-value < 0.05, |log2FC| >1). The available DNA methylation data set was limited to probes mapping to annotated active or weak promoter regions [39]. Only DEGs that showed negative correlation (adjusted p-value < 0.05, Rho < −0.6) with at least one CpG site in their promoter region were selected.
With this approach we identified 39 up-regulated and 3 down-regulated DEGs inversely correlated with methylation of corresponding promoters in ADM hypoxic tumors (Fig. 5A, Supplementary Table 6). Importantly, many of the identified genes were involved in hypoxia response (ex. MCT4/SLC16A3, CA9) including lactate metabolism (ex. LDHA, LDHB) and glycolysis (ex. P4HA2, PLOD1), indicating that the metabolic phenotype may be epigenetically driven. Other correlated genes, such as VIM, PDLIM4, and IGFBP4 are involved in epithelial to mesenchymal transition. Immunohistochemistry confirmed expression of hypoxic markers, such as SLC16A3/MCT4 and CA9 on protein level (Fig. 5B). Cluster analysis based on the identified DEGs-promoter association showed separation of three samples (Subject 36, Subject 49, and Subject 10, indicated by an asterisk in Fig. 5A) from the ADM hypoxic group (Fig. 5A). This could be explained by the heterogeneous MCT4 in two out of three samples, and for one sample the MCT4 was not available. Possibly these PanNET are truly hypoxic with a micro-vessel gradient. The hypoxic signature defined by Scarpa et al. in two of these samples was the lowest among ADM hypoxic subtype (Fig. 5A) [5]. The observed correlation between epigenetic and transcriptomic profiles of ADM hypoxic PanNETs prompted us to investigate whether they could be also discriminated on DNA methylation level. Interestingly, a principal component analysis (PCA) based on 2000 most variably methylated probes showed close clustering of MCT4 homogeneously positive samples, confirming that they also share a specific epigenetic signature (number of samples with available MCT4 immunohistochemistry = 40; Supplementary Fig. 4).
Fig. 5
Integration of methylome and transcriptome reveals changes in metabolic genes. A Hierarchical clustering of ADM PanNETs according to 42 DEGs with anti-correlated promoter DNA methylation and gene expression. Each column represents a sample, each row represents a gene. The scaled hypoxic group 3 score defined by Scarpa et al. [5] is indicated by color key (blue- low, red-high). PanNET subgroups for each sample are reported. Blue and red in the main matrix corresponds to gene expression as indicated by color key. The asterisks indicate three ADM-hypoxic samples that cluster separately. B Immunohistochemistry of MCT4 on representative samples. Each column shows one of the distinct staining patterns used in scoring. The red square delimitates area that is magnified below
Finally, analysis of DEG-probes correlation rendered remarkably fewer genes for the rest of ADM groups. Indeed, only five up-regulated DEGs were correlated with promoter methylation in ADM-NST (NR0B2, CDKN1C, C2CD4B, FERMT1, CASR) and two in ADM-immunosuppressive (GFI1 and HSPA1A) (Supplementary Tables 7 and 8). Resolving the interplay of epigenetics and gene expression in these groups will require additional studies.
Comments (0)