
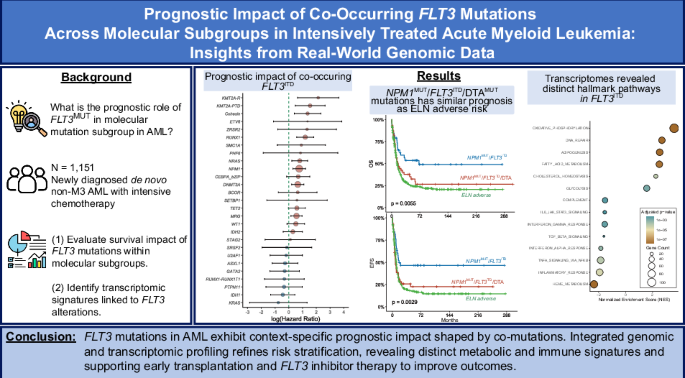
Remember me
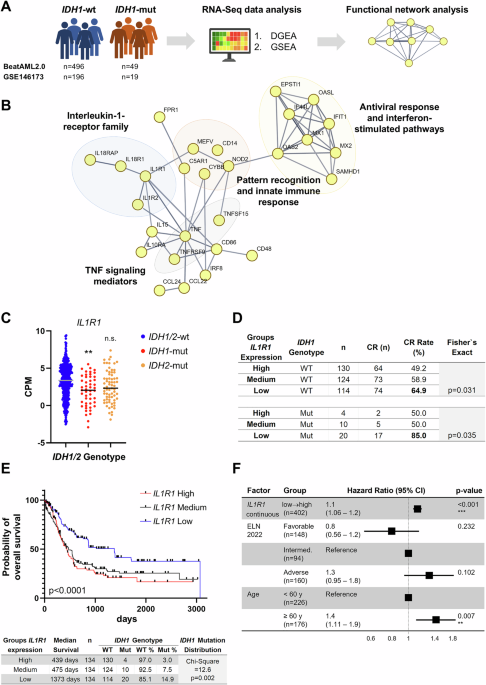
Transcriptomic profiles from 92 patients with FLT3ITD were compared to those of 335 patients with FLT3WT AML. Principal component analysis (PCA) demonstrated clear separation of clustering for transcriptomes between the two groups (Fig. 2A). DEG analysis identified 311 significantly upregulated and 536 downregulated genes, based on thresholds of |logFC | >0.585 and p < 0.05. Notably, the most upregulated genes included members of the HOX gene family, including HOXB-AS3 (logFC = 3.78), HOXB6 (logFC = 2.78), HOXB3 (logFC = 2.77), HOXA9 (logFC = 2.52), whereas TRH (logFC = −2.65) and LTF (logFC = −1.99) were among the most downregulated genes (Fig. 2B, C). GSEA revealed that FLT3ITD AML was significantly enriched for metabolic and genomic maintenance pathways, including oxidative phosphorylation (NES = 3.60, FDR < 0.001), DNA repair (NES = 2.69, FDR < 0.001), and fatty acid metabolism (NES = 2.50, FDR < 0.001; Fig. 2D), alongside with suppression of heme metabolism (NES = −2.34, FDR < 0.001; Fig. 2E). These findings highlight a metabolic reprogramming phenotype characterized by reduced heme biosynthesis and enhanced mitochondrial and DNA repair activity. To account for the known effect of NPM1MUT on HOX gene expression [11], we repeated the analysis excluding NPM1MUT cases, which yielded consistent findings (Fig. S12A–G).
Fig. 2: Transcriptomic profiling of FLT3ITD versus FLT3WT acute myeloid leukemia (AML).
A Principal component analysis (PCA) demonstrating distinct clustering of transcriptomic profiles according to FLT3ITD mutation status. B Volcano plot depicting differentially expressed genes (DEGs) between FLT3ITD and FLT3WT AML samples. C Heatmap of normalized gene rexpression values of the top upregulated and downregulated DEGs. D Dot plot showing hallmark pathways significantly enriched in upregulated genes in FLT3ITD AML. E Dot plot showing hallmark pathways significantly enriched in downregulated genes in FLT3ITD AML.
To validate these transcriptional signatures, rank-rank hypergeometric overlap (RRHO) analysis [12] in an independent Beat AML cohort revealed a strong concordance, with significant overlap in both up- and downregulated genes (Fig. S13A–C). Pathway enrichment of confirmed concordant activation of mitochondrial and metabolic programs, such as oxidative phosphorylation (NES = 1.34, FDR = 0.04), DNA repair (NES = 1.86, FDR < 0.001), and fatty acid metabolism (NES = 1.62, FDR = 0.002; Fig. S13D). Conversely, concordantly downregulated genes were enriched for immune-related programs, including interferon alpha response (NES = −3.17, FDR < 0.001), interferon gamma response (NES = −3.34, FDR < 0.001), and inflammatory response (NES = −3.14, FDR < 0.001; Fig. S13E). Together, these results provide cross-cohort validation of a reproducible FLT3ITD associated transcriptional program characterized by metabolic activation and immunologic suppression, underscoring a conserved FLT3ITD–driven phenotype.
The strengths of this study include a large, well-annotated cohort, integrated genomic-transcriptomic profiling, and validation in an external dataset. Limitations include its retrospective design, though rigorous statistical adjustments and external validation were applied. Targeted NGS limited coverage of certain FLT3 regions, potentially underestimating rare noncanonical variants. Additionally, limited FLT3 inhibitor exposure—due to recent reimbursement of midostaurin (2020) and gilteritinib (2023) in Taiwan—was addressed using multivariable models adjusting for treatment use.
In conclusion, this comprehensive genomic and transcriptomic analysis of de novo AML, we demonstrate that the prognostic impact of FLT3MUT is context-specific. FLT3ITD was associated with inferior outcomes in several molecular subgroups, while FLT3TKD showed adverse effects mainly with cohesin mutations. An FLT3ITD-driven transcriptomic signature, marked by metabolic activation and immune suppression, was externally validated. These findings refine AML risk stratification and highlight the importance of integrated genomic-transcriptomic profiling for precision therapy.
Comments (0)