
Remember me
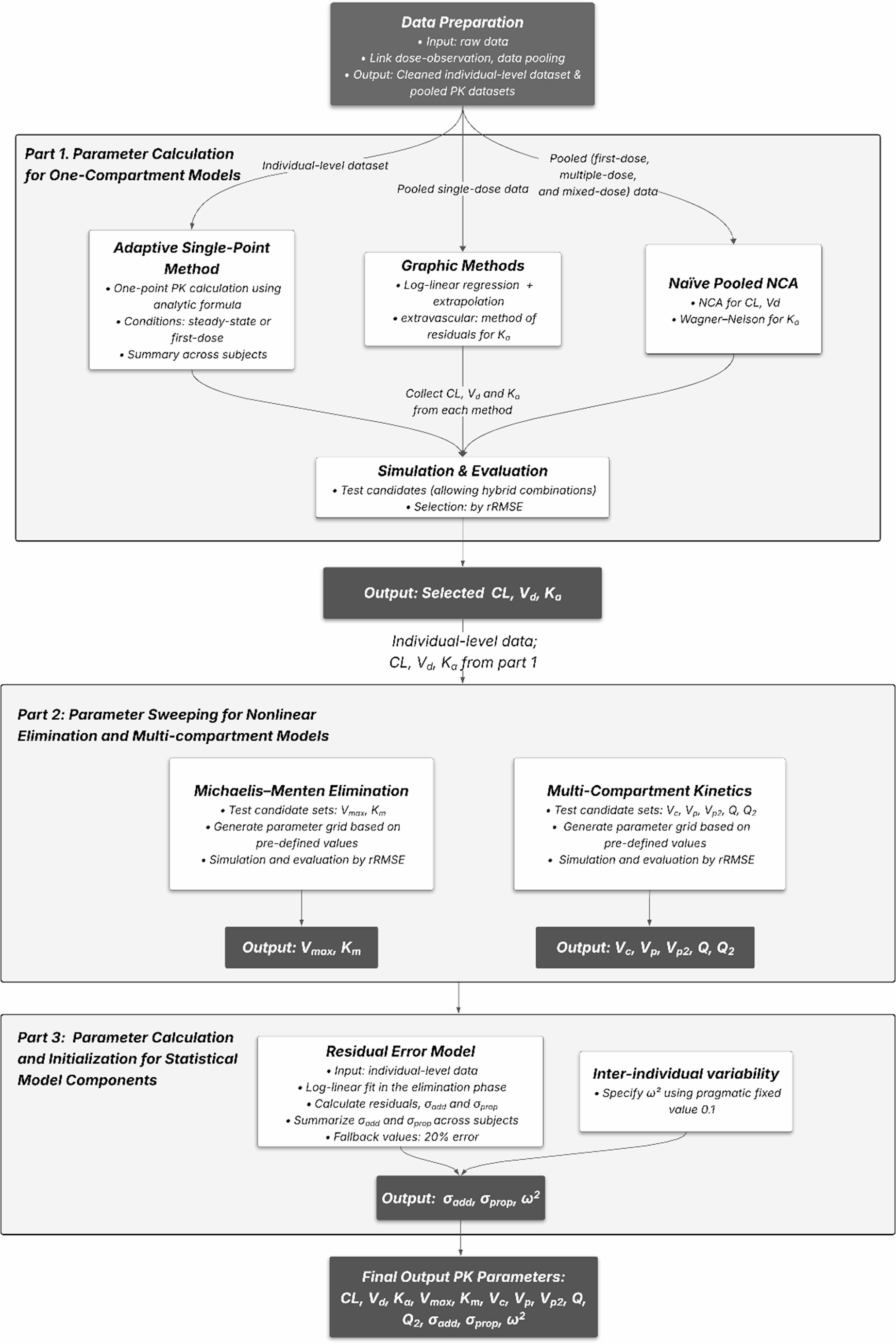
The pipeline began by processing observation records to assign dosing information, identify administration routes (bolus, infusion or extravascular), and calculate time after the last dose (TAD) (Supplementary Fig. 1, Material 1). The resulting data, hereafter referred to as individual-level data, served as the foundation for adaptive single-dose methods, parameter sweeping and residual error estimation.
A naïve pooling approach was then applied to process concentration-time data for subsequent estimation of elimination half-life (hereafter “half-life”) and analyses using NCA and graphic methods. Pooling was based on three groups: first-dose data, non-first-dose data (considered to be multiple-dose data), and mixed-dose data that included both types of dosing occasions. All concentration-time data within each group were binned and pooled based on TAD, using predefined time windows with a default number of 10, considering that adequate PK analysis typically requires 3–4 points after peak time (Tmax), and 2 points before Tmax for extravascular formulations [20, 21]. These intervals were generated by dividing unique time points into quantiles, with each group containing an approximately equal number of time points. If fewer than ten unique time points were available, the intervals were adjusted to match the actual number. Within each time window, the median time and drug concentration were calculated for each group, serving as representative values for time and concentration within that time window.
Pipeline development Part 1: parameter calculation for one-compartment models 1.Adaptive single-point method. The adaptive single-point method was designed as a framework for calculating PK parameters from single-point samples per individual, followed by population-level summarization. The framework was divided into two phases: the base phase, which computed parameters including CL and Vd, and the extended phase, which addressed cases where CL or Vd could not be calculated due to limited data and also included estimation of the absorption rate constant (Ka), which was not covered in the base phase. The overall workflow of this framework is shown in Supplementary Fig. 2 (Material 1).
1.1 Base phase. Post-first-dose and steady-state data were extracted from individuals. Steady state was defined as being achieved following administration of regularly spaced doses covering at least five half-lives or five doses, with dose intervals and fluctuations within ± 25% of the median. Half-life was estimated through linear regression on naïve pooled data. Vd was calculated as the ratio of the dose to the concentration observed at the first sampling point after the initial dose. This point was required to be collected within 20% of the half-life after dosing, during which concentration drops approximately 13% under linear elimination, to approximate the time-zero concentration. Maximum (Css, max) and minimum (Css, min) concentrations were extracted from the same interval under steady state, and their mean (Css, avg) was used to calculate CL (see Table 1). CL was subsequently derived solely based on the Css, max or Css, min, and this calculation was only applicable to intravenous cases. A geometric mean with a trim value of 0.05 (i.e., removing the top and bottom 2.5% of the data) was used to summarize PK parameters derived from individuals, given as a more robust alternative, resistant to outliers approach [22].
1.2 Extended phase. Missing CL or Vd in the base phase were derived using the estimated half-life in the extended phase (see Table 1). When both were missing, the central volume of distribution (Vc) was used as a substitute for Vd. Vc was estimated using a Cmax-based approach, calculated as the ratio of dose to the Cmax within 20% of the half-life following a single dose. For multiple doses, the accumulation ratio (Rac) was applied to adjust Css, max back to Cmax. Ka was calculated by solving the analytical concentration-time equations for a one-compartment pharmacokinetic model after a single or multiple doses. Concentration data from the absorption phase (individual sampling points at sampling times ≤ peak time) were used. CL and Vd in the equations were obtained from previous steps, and bioavailability was assumed to be 1. Ka was subsequently determined within a wide range of values (0-1000) using Brent’s method implemented in R’s uniroot function [23]. Ka and Vc were summarized by calculating the trimmed geometric mean of individual values.
Table 1 Available methods for pipeline one-compartment pharmacokinetic calculations 2.Naïve pooled NCA. Naïve pooled normalized concentration-by-dose data were used. The area under the curve (AUC) was calculated using the “linear-up log-down” trapezoidal rule. The elimination rate constant (Ke) was determined by using a best-fit strategy [24] based on log-linear regression of the terminal phase. For single-dose data, AUC from time 0 to infinity (AUC0-∞) was used for CL calculation, while for multiple-dose data, AUC0-τ was applied where τ was defined as the most commonly used dosing interval determined by frequency of administration (see Table 1). CL was calculated by dividing the dose (standardized to 1) by the AUC, and the volume of distribution of the terminal phase (Vz) was calculated using the formula Vz = CL/Ke. For the extravascular case, Ka was estimated by the Wagner-Nelson method [19]. The cumulative drug exposure at time AUC0-t was calculated, and a linear regression analysis on the fraction of the drug that remained unabsorbed during the absorption phase was performed to determine the Ka. A detailed workflow of the naïve pooled NCA process is provided in Supplementary Fig. 3 (Material 1).
3.Graphic methods. Naïve pooled first-dose data were used for this analysis. The plasma drug concentration versus time data was first plotted on a semi-logarithmic scale. Linear regression was performed on the terminal elimination phase, and the slope was used to estimate Ke, from which the half-life (t1/2) was derived. In the case of intravenous administration, the intercept, extrapolated to the y-axis, was used to calculate Vd. For extravascular administration, the method of residuals was employed to determine the Ka. This involved identifying the terminal elimination phase and subtracting it from the total plasma concentration-time curve, leaving the residuals corresponding to the absorption phase. The semi-logarithmic plot of these residuals was then used to calculate Ka. Detailed equations and workflow are listed in Table 1 and Supplementary Fig. 4 (Material 1) separately.
Part 1 evaluation and selection. Only one set of parameters was selected as final recommendations for one-compartmental parameters based on their predictive performance in a one-compartment linear model. The pipeline also allowed a hybrid combination, where each parameter (i.g., CL, Vd and Ka) could be selected independently from different methods. The predictive performance was examined through rRMSE [25,26,27], as shown in the following equation:
$$\begin\:rRMSE\:\left[\%\right]\:\:=\:100\:\times\:\\\sqrt^n\left(\frac_i-_i\right)^2}_i+_i}2\right)^2}\right)}\end$$
Where i refers to the time point, n is the total number of time points.
The set with the lowest rRMSE was selected as pipeline initial estimate recommendations for one-compartment nonlinear mixed-effects modeling and utilized to inform parameter sweeping in Part 2 of the pipeline.
Pipeline development Part 2: parameter sweeping for extended pharmacokinetic modelsIn addition to the basic one-compartment model, this pipeline provided initial estimates for extended pharmacokinetic models, including nonlinear elimination (modeled as Michaelis–Menten kinetics) and multi-compartment structures. During parameter sweeping, it defined parameter ranges, constructed a parameter grid and systematically simulated each combination using individual-level data to identify the best-fitting parameters.
Michaelis–Menten elimination. This pipeline provided initial estimate recommendations for the maximum elimination rate (Vmax), and Michaelis constant (Km), needed for nonlinear elimination modeling through parameter sweeping. This process involved a series of simulations using predefined parameter values based on a one-compartment model with Michaelis-Menten elimination, which generated simulated concentration profiles according to the dose and sampling events from input datasets, as illustrated in Fig. 2A. Parameters for simulation were categorized into test parameters (Vmax and Km) and non-test parameters (Vd and Ka). Non-test parameters (Vd) were fixed based on values obtained from Part 1. The test range for Km was scaled relative to Cmax, covering ratios from 4:1 to 1:20. Vmax was then calculated based on the Michaelis-Menten kinetic equation:
$$\:CL\:=\:\frac_}_\:+\:C}$$
Fig. 2
Example simulation outputs from a parameter sweep exploring different Km/Cmax ratios (A) and Vc/Vp ratios (B). Panel A shows simulation results using Km/Cmax ratios ranging from 4:1 to 1:20, modeled using a one-compartment model with Michaelis-Menten elimination. Panel B presents outputs for Vc/Vp ratios ranging from 10:1 to 1:10, simulated in a two-compartment model. The dose event was set as a single intravenous administration of 100 mg. Input parameters included CL = 4 L/h and Vc = 70 L, with Cmax = 100 ng/mL. In this example, C was set as 10% of Cmax for Vmax calculation in Panel A, and Q was set equal to CL in Panel B. Other values were also examined during the actual parameter sweeping, including Vmax calculated at 5%, 10%, 25%, 50%, and 75% of Cmax, and Q scaled to 0.25-, 0.5-, 1- and 2-fold of CL
Where concentration (C) was tested at 0.05, 0.1, 0.25, 0.5, and 0.75 times Cmax, and CL values were taken from Part 1. Through this battery of simulations, the parameters that provided the best-fit performance, measured by rRMSE, were identified as pipeline output.
Multi-compartmental kinetics. A similar parameter sweeping was applied to explore the volumes of distribution (Vc, Vp, and Vp2) and inter-compartmental clearances (Q and Q2). The simulated concentration profiles were generated using a two- or three-compartment model with first-order kinetics and predefined parameter values. Among these, Ka, CL, and Vc were considered non-test parameters, with values obtained from the outputs in Part 1. There were two candidate values for Vc: one from Vd (calculated through single-point, NCA, or graphic methods) and the other from Vc (output from adaptive single-point extended phase).
To construct a parameter grid for parameter sweeping, Vp was calculated based on a predefined range of Vc-to-Vp ratios, covering 10:1, 5:1, 2:1, 1:1, 1:2, 1:5, and 1:10. For three-compartment models, Vp2 was calculated using the same set. Q and Q2, were scaled relative to CL, with four candidate values tested: 0.25, 0.5, 1 and 2-fold of CL, where CL was from Part 1. Simulations were then conducted for each combination within the full parameter grid once the test spaces were determined. Figure 2B illustrates an example of such a simulation using a two-compartment model, where different Vc/Vp ratios were tested to predict concentration profiles. The most appropriate set of parameters was selected based on the rRMSE.
Pipeline development Part 3: parameter calculation and initialization for statistical model componentsInitial estimates for IIV were specified by assigning a pragmatic fixed value of 0.1 for ω2. RUV was characterized by using one of two approaches: (1) a data-driven approach using log-linear regression of terminal-phase data, with residuals computed in the original concentration scale, or (2) a fixed-fraction approach in which an expected observation error was assumed to approximate residual variability. In the data-driven approach, a linear regression was applied to the log-transformed concentration-time data from the terminal elimination phase for each subject. By default, the last three concentration-time points were used to minimize the influence of absorption and distribution. The predicted concentration at time t, denoted as \(\:_\), was obtained by back-transforming the fitted values:
$$\:_=\text\text\text(_-_\bullet\:t)$$
where \(\:_\) is the intercept and \(\:_\) is the elimination rate equal to the negative slope of the regression line.
Residuals were calculated as the difference between the observed concentrations \(\:_\) and predicted concentrations \(\:_\). The additive and proportional residual variances were then computed as follows:
$$\:_=\sqrt_-_)}\:\:\:\:\:\:_=\:\sqrt_}_}-\:1\right)}$$
where \(\:_\) represents the estimated standard deviation of additive residual error and \(\:_\) refers to the estimated standard deviation of proportional residual error.
After obtaining the individual-subject estimated \(\:\sigma\:\), a trimmed mean with a trimming proportion of 0.05 was applied to summarize the results, thereby reducing the influence of outliers. For the fixed-fraction method, when data were insufficient for data-driven estimation, empirical default values were used. According to the NONMEM User Guides [28], initial standard deviations could be set as a fraction (e.g., 20%) of the typical observed value, as shown in the following equation:
$$\:_=\:CV\%\:\times\:\stackrel$$
where \(\:\stackrel\) represents the average observed concentration across the entire dataset, and CV% refers to the expected value for percentage error of the observations. The default setting in the pipeline was 20%.
DataA total of 21 simulated and 13 real-life datasets were analyzed. The simulated datasets comprised seven intravenous bolus, seven intravenous infusion, and seven oral cases, each of which included four rich, one semi-sparse, and two sparse designs. The pharmacokinetic models included 12 one-compartment linear, three one-compartment nonlinear (with Michaelis-Menten elimination), three two-compartment linear, and three two-compartment nonlinear models. The 13 real-life datasets included eight intravenous and five oral administration profiles, with five rich and eight sparse sampling designs.
Simulated data. All simulated datasets are provided in the supplementary material. 15 out of 21 datasets were obtained directly from the nlmixr2data package [29]. Additionally, three rich one-compartment datasets, Bolus_1CPT, Infusion_1CPT, and Oral_1CPT from nlmixr2data, were extended by generating semi-sparse, sparse1, and sparse2 datasets for each, respectively. The semi-sparse dataset was created by dividing the original IDs into three groups, where each group included only two sampling points within a single dosing interval following multiple doses. The sampling points differed among the groups at post-dose 2, 4, 6, 8, 12, and 24 h. Sparse1 datasets had two or three sampling points available in a different dose interval for all IDs after multiple doses, with sampling time after the last dose at 2 (if oral), 20, and 24 h. Sparse2 datasets had two or three data points collected at 2 (if oral), 20, and 24 h after the single dose.
Real-life data. Real-life data consisted of three datasets, theo_sd, theo_md, and pheno_sd sourced from nlmixr2data [29], as well as ten datasets from nine published articles. Information about these ten datasets is detailed in Supplementary Table 1 (Material 2). The concentration-time curves for the simulated and real-life datasets were provided in Supplementary Figs. 1 and 2 (Material 2).
Pipeline performanceFor the simulated dataset, the pipeline was evaluated by re-estimating the simulated cases using nonlinear mixed-effects modeling, applying the true model that generated the data and the initial estimates proposed by the pipeline. Accuracy was defined as the deviation (%), calculated as the absolute relative difference between the final estimate and the true value used in the simulation.
$$\:Deviations\:\left(\%\right)=\left|\frac\:-\:_}_}\right|\:\times\:100\%$$
where \(\:\widehat\) is the final parameter estimate, and \(\:_\) is the true value of the parameter used for simulation.
A threshold of 20%, as an often-used clinical relevance threshold [28, 30], was applied to evaluate whether the final estimates recovered the true values. For each parameter, the proportion of datasets was calculated in which the absolute relative deviation between the final estimate and the true value did not exceed 20%. As an exploratory analysis, a 30% threshold, also commonly used as a reference for determining clinical relevance in practice [31, 32] was used for evaluation. Overall success rates were computed under both thresholds, defined as the proportion of datasets in which all parameters simultaneously met the respective criterion. The pipeline performance was also compared with the following initial estimate designs for simulated datasets. These strategies were:
1.Setting all initial estimates to 1 before back-transformation (expressed as inits = 1 in the following description), with parameters defined using log-transformation. For example, the initial estimate of CL was specified to 1, which corresponds to setting the log-transformed CL to 1 in the initial condition function.
2.Setting all estimates to 1 before back-transformation, followed by optimizing the initial estimates using optimization methods available in the R statistical environment [33]: nls (Gauss-Newton method), nlm (Newton-type method), and nlminb (quasi-Newton method), (expressed as inits = nls, inits = nlm, inits = nlminb in the following description) through compartmental analysis without considering IIV.
For real-life clinical data, where the true model structure and parameter values were unknown, parameter estimation was conducted using one- and two-compartment models with IIV on all parameters and a combined residual error model. Model performance using the pipeline and the inits = 1 strategy was then compared. The evaluation focused on assessing the precision of the final parameter estimates obtained using both strategies, as well as the model’s goodness-of-fit, measured by AIC, and computation time. Stochastic approximation expectation-maximization (SAEM) and first-order conditional estimation with interaction (FOCEI) algorithms were used for test work in simulated and real-life datasets.
SoftwareThe pipeline was developed in R and is available as an R package, nlmixr2autoinit (version 1.0). The code is available on GitHub (https://github.com/ucl-pharmacometrics/nlmixr2autoinit). The nlmixr2 package was used for model parameter estimation.
Comments (0)