
Remember me

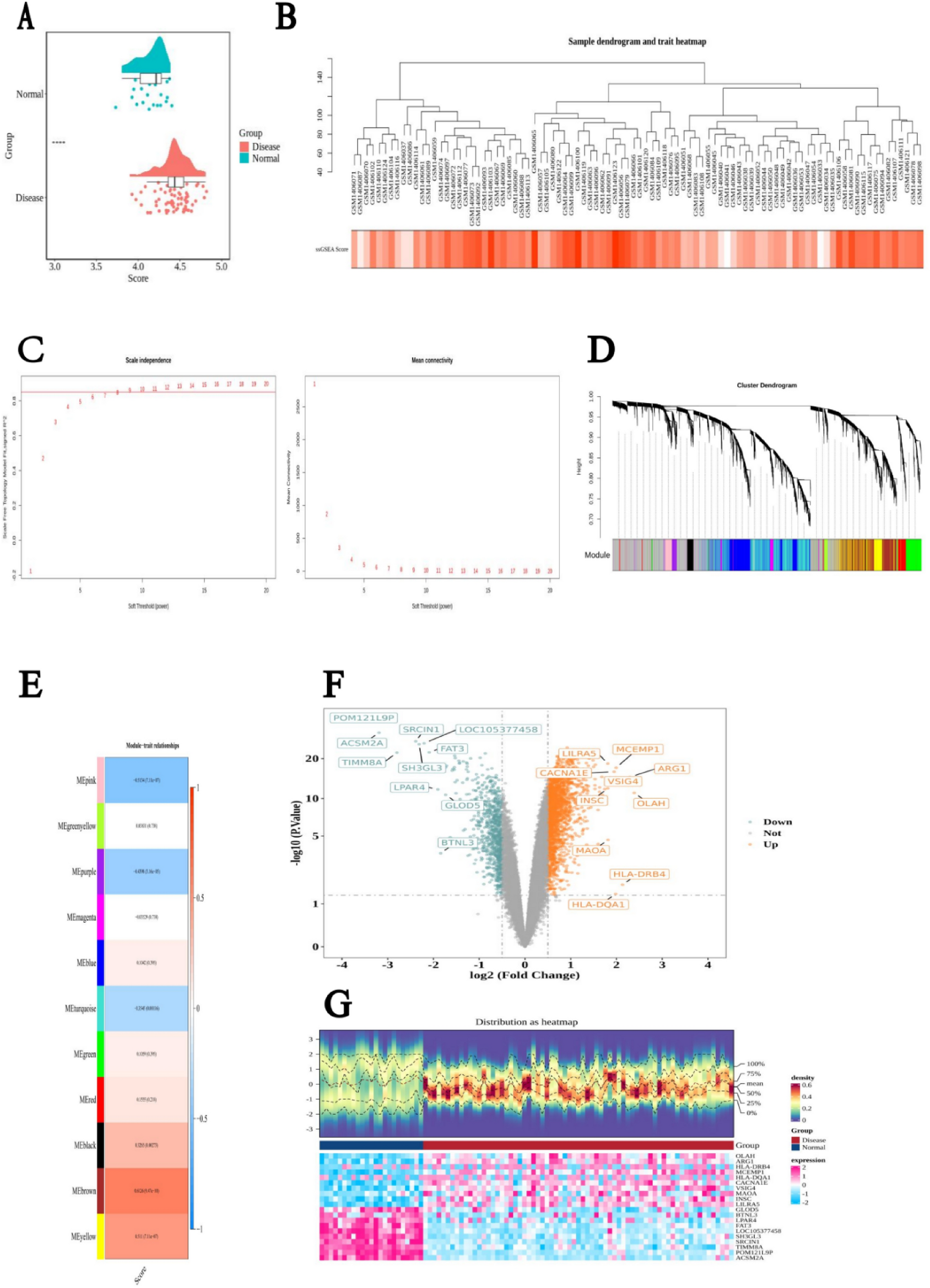

In the GSE58294 dataset, significant differences in the CMRGs score were observed between disease and normal samples (P < 0.0001), with disease samples showing a higher CMRGs score.
For WGCNA, no outlier samples were detected in the GSE58294 dataset. The optimal power value was determined to be 8, as this produced an R2 value greater than 0.85 (indicated by the red line) and resulted in mean connectivity close to 0. The co-expression matrix was then used to merge similar modules, identifying 11 gene modules (excluding the gray module). The Mebrown module showed a strong positive association with the CMRGs score (cor = 0.6126, P = 9.14 × 10−10), while the Mepink module showed a strong negative association with the CMRGs score (cor = − 0.5134, P = 7.11 × 10–7). Therefore, the 1037 genes from the combination of these two key modules were identified as key module genes.
Additionally, in the GSE58294 dataset, a total of 2783 DEGs were identified, with 1969 genes up-regulated and 814 genes down-regulated in IS samples (Fig. 1).
Fig. 1
Module gene selection, ssGSEA scoring, sample clustering, and soft-threshold analysis. A Differential analysis of ssGSEA scores between IS and normal samples. B Sample hierarchical clustering based on the expression matrix of all samples in the training set (GSE58294), followed by weighted gene co-expression network analysis (WGCNA). C Determination of the optimal soft-threshold power. The left panel shows the scale-free topology model fit index under different soft-thresholds, while the right panel illustrates the mean connectivity across thresholds. D Dynamic tree cut for module detection. The upper part shows the gene hierarchical clustering dendrogram, and the lower part indicates the identified gene modules. E Heatmap of correlations between gene modules and clinical traits. The colored bars on the left represent module assignments, and the color scale on the right indicates the strength of correlation. F Volcano plot of differentially expressed genes (DEGs) between the Disease and Normal groups in the training set. Green dots indicate downregulated genes, and orange dots indicate upregulated genes. G Heatmap of DEG expression profiles in the training set. The top panel shows sample-wise expression distribution; the lower heatmap depicts expression values of individual genes across samples
Determination and function exploration of candidate genesA total of 336 candidate genes were pinpointed by intersecting 1037 key module genes with 2783 DEGs. These candidate genes were significantly enriched in 487 GO terms (P < 0.05), including 362 BPs, 67 CCs, and 58 MFs. Notably, the enriched GO terms included "cellular cation homeostasis" (BP), "secretory granule membrane" (CC), and "phospholipid binding" (MF) (Supplementary Table S2). Furthermore, the candidate genes were significantly enriched in 17 KEGG pathways (P < 0.05), such as "cytokine-cytokine receptor interaction", "chemokine signaling pathway, and "B cell receptor signaling pathway"(Supplementary Table S2).
To further investigate the interactions among these candidate genes, a PPI network was constructed, consisting of 97 genes and 89 relationships, with 239 outlier genes removed. For example, ZAP70 exhibited strong interactions with several other genes, including PLGG1 and IL2RB, indicating that ZAP70 might serve as a key regulatory hub within the network (Fig. 2).
Fig. 2
Identification of candidate genes and functional enrichment analysis (GO, KEGG) and PPI network construction. A Venn diagram showing the identification of candidate genes, with a total of 336 genes obtained. B GO enrichment analysis. The left panel displays a circular plot of enriched GO terms, and the right panel provides a detailed description of the corresponding biological processes. C KEGG pathway enrichment analysis illustrating interactions and relationships among pathways through color-coded lines and labels. D Protein–protein interaction (PPI) network. Blue circles represent the 97 candidate genes involved in protein interaction networks
Revelation of Robust Causal Relationships Between 12 Significant Genes and ISPerforming MR analysis with candidate genes as exposure factors revealed that 12 genes exhibited significant causal relationships with IS (PIVW < 0.05) (Table 1). These genes were: CXCL16, ST3GAL4, ACTA2, WWC3, NECAB2, FLOT1, INTS3, NLRX1, STRBP, SMG5, DHRS13, ABHD4. Of these, 9 genes were linked to an increased risk of IS (OR > 1, 95% Confidence Interval [CI] ≠ 1, PIVW < 0.05), while the remaining 3 genes were identified as protective (OR < 1, 95% CI ≠ 1, PIVW < 0.05). These associations were visually confirmed through scatter plots, where risk-associated exposure factors showed positive regression slopes, while protective exposure factors demonstrated negative slopes (Supplementary Fig. S1). Forest plot analysis employing the IVW method consistently revealed positive effect sizes for risk-associated exposure factors (MR effect sizes > 0) and negative effect sizes for protective exposure factors (MR effect sizes < 0) (Supplementary Fig. S2). Funnel plots further supported the findings, showing a symmetric distribution of IVs around the IVW line for each exposure factor, which adhered to Mendel’s second law. (Supplementary Fig. S3).
Table 1 The results of MR studyNext, a heterogeneity test revealed that the P values for all 12 genes were greater than 0.05 (fixed IVW method was applied), suggesting no significant heterogeneity (Supplementary Table S3). Additionally, the horizontal pleiotropy test showed P values of all 12 genes greater than 0.05, suggesting the absence of horizontal pleiotropy (Supplementary Table S4). LOO analysis confirmed the robustness of the MR results, with no significant deviations (Supplementary Fig. S4). Finally, the Steiger test revealed that the causal directions for 12 genes were correct (SNP r2.exposure > SNP r2.outcome, P < 0.05), further supporting the reliability of the MR findings (Supplementary Table S5).
In conclusion, based on the combined results from the MR study, sensitivity analyses, and the Steiger test, the 12 identified genes were deemed as significant genes for further investigation.
Identification and Examination of CXCL16, ST3GAL4, ACTA2 as Biomarkers for ISFrom an initial set of 12 significant genes, 10 candidate biomarkers (WWC3, NECAB2, ACTA2, ST3GAL4, FLOT1, INTS3, NLRX1, CXCL16, STRBP, and SMG5) were identified through LASSO analysis (lambda.min = 0.0255). Subsequent gene expression analysis revealed that CXCL16, ST3GAL4, and ACTA2 were significantly up-regulated in disease samples from both the training (GSE58294) and validation (GSE16561) datasets (P < 0.05). Notably, INTS3, NLRX1, and NECAB2 were not detected in the GSE16561 dataset. Therefore, CXCL16, ST3GAL4, and ACTA2 were selected as biomarkers for IS, which could be valuable for early diagnosis and understanding the underlying mechanisms of the disease. Additionally, ROC curve analysis showed AUC values greater than 0.80 for all biomarkers, indicating their excellent diagnostic ability.
Correlation analysis revealed significant positive correlations among all biomarkers. For instance, CXCL16 exhibited the strongest positive correlation with ST3GAL4 (cor = 0.52, P < 0.001). Functional similarity analysis further demonstrated that CXCL16 had the strongest functional similarity (Fig. 3).
Fig. 3
Biomarker selection, correlation analysis, and functional similarity (Friend) analysis. A LASSO regression for prognostic gene selection. The optimal value of the lambda parameter is determined at the point with the minimum mean squared error on the curve. B Boxplots of candidate biomarker expression levels. The left panel shows expression in the training set, and the right panel shows expression in the validation set. C ROC curves of signature genes in the training set. The bottom-right legend lists each candidate biomarker along with its corresponding AUC value. D Correlation analysis of biomarkers in the IS training set, indicating that CXCL16 exhibits relatively strong functional similarity with other biomarkers. E Boxplot of functional similarity (Friend analysis) among biomarkers. Red represents ACTA2, green represents ST3GAL4, and blue represents CXCL16
Establishment of Well-Performing NomogramA nomogram model was developed based on the identified biomarkers, with specific point values assigned to each gene. Higher total points in the model were associated with an increased risk of IS. The calibration curve confirmed the model’s accuracy, with a P value of 0.412 in the HL test. The decision curve demonstrated that the nomogram provided a greater net benefit compared to single factors, indicating its excellent predictive ability. The CIC showed that the model’s predictions closely aligned with actual occurrences, further confirming the clinical utility of the nomogram. ROC analysis revealed an AUC of 0.945 for the nomogram model, highlighting its strong predictive capability. Overall, these results underscored the robust predictive efficacy of the nomogram in assessing IS risk (Fig. 4).
Fig. 4
Construction and evaluation of the nomogram model. A Nomogram based on selected biomarkers. The “Total Points” represents the sum of the individual scores corresponding to each gene expression level, which can be mapped to the predicted probability of IS at the bottom of the figure. B Calibration curve of the nomogram. The Hosmer–Lemeshow test yielded a p-value > 0.05, indicating no significant difference between predicted and actual outcomes, and a good model fit. The mean absolute error (MAE) < 0.1 suggests minimal deviation between predicted and observed risk, supporting high predictive accuracy. C Decision curve analysis (DCA) for the biomarkers CXCL16, ST3GAL4, and ACTA2. All regression models showed positive net benefit, indicating that the nomogram provides good clinical utility. D Clinical impact curve (CIC). The CIC demonstrates that as the threshold probability increases, the number of predicted high-risk patients closely matches the actual cases, indicating improved clinical prediction efficiency. E ROC curve of the nomogram model, suggesting a certain diagnostic value for predicting IS
Investigation of Functions and Pathways of BiomarkersGSEA analysis revealed that CXCL16 was significantly enriched in 67 pathways, ST3GAL4 in 54 pathways, and ACTA2 in 55 pathways (|NES|> 1, P < 0.05, and q < 0.05) (Supplementary Table S6). The top 5 enriched pathways (ranked by P values from lowest to highest) for CXCL16, ST3GAL4, and ACTA2 were visualized. Notably, several co-enriched pathways for all three biomarkers, such as "lysosome" and "toll-like receptor signaling pathway", might play crucial roles in the pathogenesis of IS. The co-enrichment of the lysosome and toll-like receptor signaling pathways in the context of IS suggested a complex interplay between cellular stress response and immune regulation.
Additionally, GSVA analysis indicated that CXCL16 was enriched in 90 pathways, ST3GAL4 in 90 pathways, and ACTA2 in 64 pathways (|t|> 2 and P < 0.05) (Supplementary Table S7). The top 10 activated pathways (t > 2) and the top 10 inhibited pathways (t < − 2) for each of the biomarkers were displayed in Fig. 5D–F. For example, co-enriched activated pathways included "galactose metabolism", while the co-enriched inhibited pathway comprised "basal transcription factors". These findings suggested that the identified pathways were involved in critical biological processes that might contribute to the development and progression of IS (Fig. 5).
Fig. 5
GSEA and GSVA enrichment analyses of prognostic genes. A GSEA results for CXCL16, showing enrichment of CXCL16-related genes in 67 pathways. B GSEA results for ST3GAL4, with 54 pathways enriched by ST3GAL4-associated genes. C GSEA results for ACTA2, identifying 55 enriched pathways related to ACTA2. D GSVA enrichment analysis of CXCL16, primarily enriched in 90 KEGG pathways including KEGG_MISMATCH_REPAIR. E GSVA enrichment analysis of ST3GAL4, primarily enriched in 90 KEGG pathways including KEGG_PRIMARY_IMMUNODEFICIENCY. F GSVA enrichment analysis of ACTA2, mainly enriched in 64 KEGG pathways including KEGG_BUTANOATE_METABOLISM
Exploration of Immune Infiltration Differences Between IS Disease and Normal SamplesA stacked chart illustrated the immune infiltration scores of 28 immune cell types in both IS disease and normal samples. Further analysis of immune cell infiltration revealed significant differences in the levels of 16 immune cell types between IS disease and normal samples (P < 0.05). For instance, activated B cells showed significantly lower infiltration in IS disease samples (P < 0.0001), while activated dendritic cells exhibited notably higher infiltration in IS disease samples (P < 0.01). Correlation analysis revealed a strong positive correlation between immature B cells and activated B cells (cor = 0.75, P < 0.001), while activated B cells displayed a significant negative correlation with macrophages (cor = -0.70, P < 0.001). Additionally, the correlation analysis further revealed that activated B cells and effector memory CD4 T cells were significantly negatively correlated with all biomarkers (cor < -0.30, P < 0.001), while activated dendritic cells, immature dendritic cells, macrophages, myeloid-derived suppressor cells (MDSC), neutrophils, and regulatory T cells showed significant positive correlations with all biomarkers (cor > 0.30, P < 0.05) (Supplementary Table S8) (Fig. 6).
Fig. 6
Immune cell infiltration analysis in Disease and Normal samples. A Heatmap of immune cell abundance estimated by ssGSEA, showing 28 types of immune cells across all samples. B Differential analysis of immune cell infiltration between Disease and Normal groups, identifying 16 significantly altered immune cell types, including Activated B cells. C Correlation heatmap of differentially expressed immune cells. A strong positive correlation was observed between Immature B cells and Activated B cells (r = 0.86, p < 0.05), while Macrophages and Activated B cells showed a significant negative correlation (r = –0.70, p < 0.05). D Heatmap of correlations between biomarkers and differentially expressed immune cells. All three biomarkers were negatively correlated with Activated B cells and Effector memory CD4 T cells. Additionally, ST3GAL4 showed a positive correlation with Memory B cells, while ACTA2 was negatively correlated with Natural killer cells
Revealing the Potential Molecular Mechanisms of BiomarkersBy searching, 12 key miRNAs targeting biomarkers were identified through the overlap of predictions from both databases. Among them, 1 key miRNA (hsa-mir-587) targeted CXCL16, 6 key miRNAs (eg. hsa-mir-220b, hsa-mir-197-3p) targeted ST3GAL4, and 5 key miRNAs (eg. hsa-mir-27b-3p, hsa-mir-181d-5p) targeted ACTA2 (Supplementary Fig. S5). Subsequently, 203 lncRNAs (eg. SNHG1, FTX) targeting these key miRNAs were identified, and a lncRNA-miRNA-mRNA network was constructed. Besides, we further identified 22 TFs targeting the biomarkers. Notably, some TFs, such as AR, MYC, and KLF4, were found to co-target multiple biomarkers. This co-targeting suggested potential coordinated regulation of these biomarkers by multiple TFs, which could be crucial for understanding their function in the cellular context.
Besides, among 100 IS-RGs, 9 genes (IL6, PLAT, BDNF, CPS1, SH2B3, FLNA, ASS1, WFS1, FGFR1) exhibited significant expression differences between IS disease and normal samples, showing the lower expression in IS disease samples (P < 0.05). Correlation analysis showed BDNF had the significant negative correlation with all biomarkers (cor < -0.30, P < 0.01) (Fig. 7).
Fig. 7
Construction of the ceRNA network, biomarker–TF regulatory network, and correlation analysis with disease-associated genes. A miRNA–mRNA regulatory network. Red nodes represent the identified biomarkers, blue nodes represent predicted miRNAs, and purple nodes represent predicted lncRNAs. B Transcription factor (TF)–biomarker regulatory network. Red nodes indicate prognostic genes, pink and purple nodes represent predicted TFs, with purple denoting TFs identified by multiple prediction methods. C Boxplot of expression levels of disease-associated genes in the IS training set. Nine disease-related genes were significantly differentially expressed. D Correlation analysis between biomarkers and disease-associated genes in the IS training set. All three biomarkers showed negative correlations with IS-related genes
Investigating the Targeted Drugs for BiomarkersIn this study, a comprehensive drug prediction approach was employed, identifying 89 potential drugs targeting the biomarkers, such as probucol, rifaximin, and formoterol. A drug-biomarker network was subsequently constructed to visualize these interactions. Based on the combined score, benzene, which exhibited the highest combined score for CXCL16, and probucol, with the highest combined score for ACTA2, were selected for detailed molecular docking studies. For ST3GAL4, Idose (rank third) was chosen for molecular docking analysis after a thorough evaluation of combining energy results.
The molecular docking results provided valuable insights into the binding affinities and interactions between the selected drugs and their respective biomarkers. Specifically, ACTA2 and probucol demonstrated a particularly strong binding energy of − 7.7 kcal/mol, facilitated by interactions at key residues such as G184, G304, and K217. Similarly, ST3GAL4 and Idose exhibited a binding energy of -5.8 kcal/mol, characterized by interactions at residues L147, T261, and H294, indicating a stable interaction that could be explored for therapeutic purposes. The binding energy between CXCL16 and benzene was -3.5 kcal/mol, suggesting a moderate interaction that warrants further investigation (Fig. 8). To further validate the reliability of the molecular docking approach, we selected known target protein inhibitors reported in the literature as positive controls for docking analysis, including: the ACTA2 inhibitor Y-27632 (Aguado et al. 2022), the CXCL16 inhibitor Vx-702 (https://www.scbt.com/browse/cxcl16-inhibitors), and the ST3GAL4 inhibitor Brigatinib (Han et al. 2024). Results indicated binding energies of − 7.4 kcal/mol for ACTA2-Y-27632, − 8.1 kcal/mol for CXCL16-Vx-702, and − 9.9 kcal/mol for ST3GAL4-Brigatinib (Fig. 9A–C). These control results aligned with expectations, confirming the reliability of the molecular docking approach employed in this study. These findings highlighted the potential of these drugs to interact with their respective biomarkers, providing a foundation for the development of targeted therapies.
Fig. 8
Drug prediction and molecular docking analysis. A Biomarker–drug interaction network. Red nodes represent biomarkers, and pink nodes represent predicted drug candidates. B–D Molecular docking models of key genes with their corresponding predicted drugs: B CXCL16 with benzene; C ST3GAL4 with Idose; D ACTA2 with probucol
Fig. 9
Molecular docking results between the proteins encoded by biomarkers and their respective inhibitors. A CXCL16 with Vx-702. B ST3GAL4 with Brigatinib. C ACTA2 with Y-27632
Comments (0)