Chemicals and Reagents
L-glutamic acid monosodium salt monohydrate (glutamate, cat. no. 49621) was obtained from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, cat. no. 11965-092), fetal bovine serum (FBS; Gibco, cat. no. 16000-044), 0.25% trypsin–EDTA (Gibco, cat. no. 25200-056), and antibiotic solution containing 10,000 U/mL penicillin and 10 mg/mL streptomycin (Gibco, cat. no. 15140-122) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Pifithrin-α (PFT-α, cat. no. EMD_BIO-506132) was obtained from Calbiochem (San Diego, CA, USA).
Cell Culture
Glutamate-sensitive HT22 murine hippocampal neuronal cells (HT22 cells, a kind gift from Dr. David Schubert, Salk Institute, La Jolla, CA, USA) and BV2 murine microglial cells (BV2 cell, ABC-TC212S; AcceGen Biotechnologies) were used in this study. Cell line authentication was performed by short tandem repeat (STR) profiling, and both cell lines matched their corresponding reference profiles. HT22 cells were used at passages 3–15, and BV2 cells were used at passages 3–10. All cell lines were authenticated and routinely tested for mycoplasma contamination using MycoStrip (InvivoGen, cat. no. rep-mys-50), and all tests were consistently negative. Cells were maintained in DMEM (Gibco, cat. no. 11965-092) supplemented with 10% (v/v) FBS (Gibco, cat. no. 16000-044) and 1% (v/v) penicillin-streptomycin (Gibco, cat. no. 15140-122). Cells were incubated at 37 °C in a humidified atmosphere containing 5% CO₂ and sub-cultured every 2–3 days or before reaching approximately 80–90% confluence.
Cell Viability Assay
For viability assays, HT22 cells were seeded in 96-well plates at a density of 5 × 10³ cells per well and allowed to adhere overnight. Cells were then treated with glutamate for 6, 12, or 24 h. For glutamate treatment, a stock solution of L-glutamate (1 M) was prepared in serum-free DMEM and diluted in culture medium immediately before use to the indicated final concentrations. Cell viability was assessed using the MTT assay. Briefly, 10 µL of MTT solution (5 mg/mL in PBS) was added to each well (final concentration: 500 µg/mL), followed by incubation for 3 h at 37 °C. The medium was carefully removed, and 100 µL of dimethyl sulfoxide (DMSO) was added to dissolve the formazan crystals. Absorbance was measured at 570 nm using a microplate reader (Synergy H1, BioTek). Relative cell viability was expressed as a percentage of the untreated control group. Blank wells containing medium only were used for background subtraction.
Pharmacological Inhibitor Treatments
Pharmacological inhibitors used in this study were prepared as concentrated stock solutions, including SP600125 (SP; 20 mM in DMSO; Abcam, cat. no. ab120065), SB203580 (SB; 20 mM in DMSO; Sigma, cat. no. S8307), U0126 (MEK1/2 inhibitor; 10 mM in DMSO; Cell Signaling Technology, cat. no. 9903), actinomycin D (ActD; 10 mg/mL; Sigma, cat. no. A9415), and cycloheximide (CHX; 100 mg/mL; Sigma, cat. no. C4859). Stock solutions were diluted to their final working concentrations in culture medium immediately before use.
Preparation of Microglia-Conditioned Medium (CM)
For preparation of microglia-conditioned medium (CM), BV2 cells were stimulated with lipopolysaccharide (LPS, 1 µg/mL; Sigma-Aldrich, cat. no. L2654) for 10 h at 37 °C. The conditioned medium was collected and centrifuged at 2,000 × g for 10 min to remove cellular debris. The clarified supernatant was then applied to HT22 neuronal cultures at final concentrations of 30%, 50%, or 100% (v/v) in fresh culture medium and incubated for 24 h prior to further analysis.
Glutamate Quantification
Extracellular glutamate levels in BV2-derived conditioned medium were quantified using the Glutamate-Glo™ Assay (Promega, cat no. J7021). BV2 cells were stimulated with LPS (1 µg/mL) for 10 h, and the resulting CM, prepared as described above, was subjected to glutamate measurement. All procedures were performed according to the manufacturer’s instructions. The clarified supernatant was transferred to a white 96-well plate, mixed with an equal volume of detection reagent, and incubated for 30 min at room temperature in the dark. Luminescence was measured using a microplate reader (Synergy H1, BioTek), and results were expressed as relative light units (RLU) normalized to the mean value of control CM (con-CM).
Primary Culture of Embryonic Mouse Hippocampal Neurons
Primary hippocampal neurons were prepared from embryonic day 18 (E18) ICR mouse embryos as previously described (Dong et al. 2009). Pregnant mice were euthanized by CO₂ asphyxiation, and embryos were aseptically removed and decapitated. Hippocampi were rapidly dissected in cold Hank’s Balanced Salt Solution (HBSS; Gibco, USA) and digested with 0.25% trypsin–EDTA (Gibco) at 37 °C for 15 min. The tissue was washed and gently dissociated using a fire-polished glass pipette in DMEM containing 10% FBS to inactivate trypsin. Neurons were counted and plated at 5 × 10⁴ cells/well on poly-D-lysine-coated (0.1 mg/mL; Sigma-Aldrich) glass coverslips in 24-well plates. Neurons were maintained in Neurobasal medium (Gibco, cat. no. 21103049) supplemented with 2% B-27 Supplement (Gibco, cat. no. 17504044), 0.5 mM GlutaMAX (Gibco, cat. no. 35050061), and 1% penicillin–streptomycin (Gibco) at 37 °C in a humidified atmosphere containing 5% CO₂. Half of the medium was replaced every 3 days, and neurons at 7 days in vitro (DIV7) were used for subsequent experiments.
Immunocytochemistry
Immunocytochemistry was performed as previously described (Dong et al. 2009; Lewerenz and Maher 2015). Cells were washed three times with PBS and fixed with 3% paraformaldehyde containing 0.1 mM CaCl₂ and 0.1 mM MgCl₂ (pH 7.4) for 10 min. After fixation, cells were permeabilized with 0.2% Triton X-100 in PBS for 5 min and blocked with 10% normal goat serum (Jackson ImmunoResearch, cat. no. 005-000-121, West Grove, PA, USA) for 1 h at room temperature. Samples were incubated overnight at 4 °C with a primary antibody against CREB2/ATF4 (mouse monoclonal clone B-3, 1:50, sc-390063; Santa Cruz Biotechnology). After three washes, cells were incubated with FITC-conjugated goat anti-mouse secondary antibody (1:200; Jackson ImmunoResearch, cat. no. 115-095-003) for 1 h. Nuclei were counterstained with 5 µM Hoechst 33,342 (stock solution diluted immediately prior to use; Thermo Fisher Scientific, Cat. No. H1399) for 5 min at room temperature. After staining, cells were washed three times with PBS, and coverslips were mounted using Vectashield mounting medium (Vector Laboratories, cat. no. H-1000).
Confocal Imaging
Fluorescence images were acquired using a confocal laser scanning microscope (AXIO series, Carl Zeiss, Oberkochen, Germany) equipped with Axiovision 4.9 software using Plan-Apochromat 20×/0.8 NA, 40×/1.3 NA oil, and 63×/1.4 NA oil-immersion objectives. Cells were initially screened at 20× magnification, and representative fields were selected for imaging. FITC-labeled CREB2 was imaged using 488-nm excitation and 500–550-nm emission, and Hoechst-stained nuclei were imaged using 405-nm excitation and 420–480-nm emission. Images were collected with a 1 Airy pinhole, a scan speed of 1–2 µs/pixel, and 2–4× line averaging at 16-bit depth, using identical gain and exposure settings across all samples.
Western Blotting
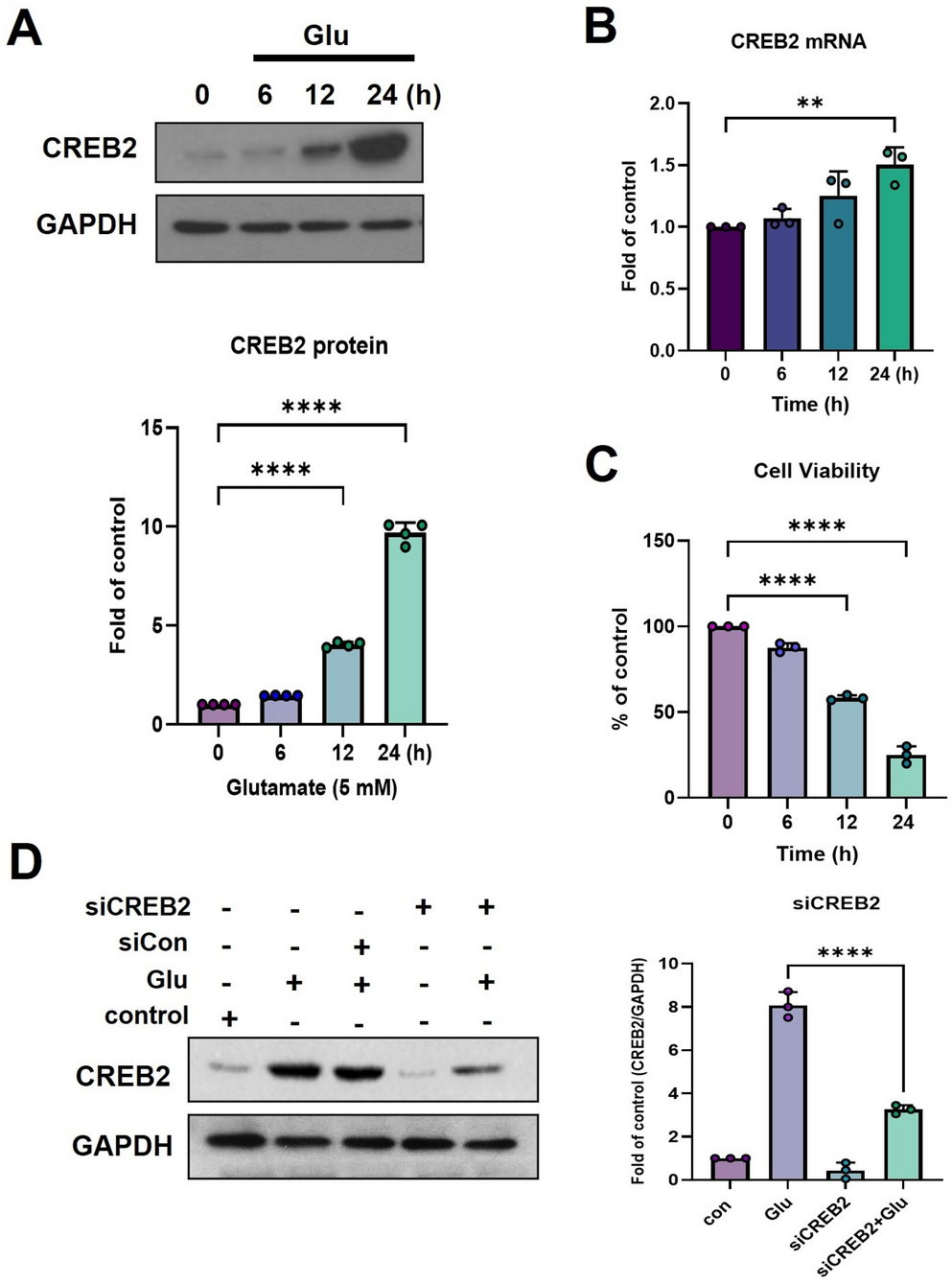
Cells were lysed in ice-cold RIPA buffer (Thermo Scientific, cat. no. 89900) supplemented with 1× dual protease/phosphatase inhibitor cocktail (100× stock; Thermo Fisher Scientific, cat. no. 78440). Lysates were sonicated using a probe sonicator (Q125, QSonica, USA) equipped with a 3-mm microtip (Part #4422) designed for cell disruption. Samples were kept in 1.5-mL microtubes placed in an ice bath, and sonication was performed at 20% amplitude for 10 s in pulse mode (1 s ON/1 s OFF) to ensure complete cell disruption while minimizing heat generation. Lysates were then clarified by centrifugation at 12,000 × g for 15 min at 4 °C. Protein concentrations were determined using the Bio-Rad Protein Assay (Bio-Rad, cat. no. 500-0006, Hercules, CA, USA). Equal amounts of protein (20–30 µg) were loaded on 10–12% SDS–polyacrylamide gels and transferred onto PVDF membranes (Bio-Rad, cat. no. 1620177). Membranes were blocked with 5% skim milk in TBS-T (20 mM Tris-HCl, 150 mM NaCl, 0.1% Tween-20) for 1 h at room temperature and subsequently incubated overnight at 4 °C with primary antibodies against CREB2/ATF4 (mouse monoclonal, clone B-3, 1:100; Santa Cruz Biotechnology, cat. no. sc-390063, RRID: AB_10917316), p53 (1:1000; Cell Signaling Technology (CST), cat. no. 12571, RRID: AB_27113937), phospho-p53 (Ser15) (1:1000; CST, cat. no. 9284, RRID: AB_331464), GADD45α (1:1000; CST, cat. no. 4632, RRID: AB_10694428), GAPDH (1:1000; CST, cat. no. 5174, RRID: AB_10622025), and β-actin (1:200; Santa Cruz Biotechnology, cat. no. sc-47778, RRID: AB_2714189). After washing with TBS-T, membranes were incubated with HRP-conjugated anti-mouse IgG (1:5000; CST, cat. no. 7076, RRID: AB_330924) or HRP-conjugated anti-rabbit IgG (1:5000; CST, cat. no. 7074, RRID: AB_2099233) secondary antibodies for 1 h at room temperature. ECL chemiluminescent substrate (Amersham ECL Western Blotting Detection Reagent, GE Healthcare, cat. no. RPN2109) and imaged using a ChemiDoc imaging system (Bio-Rad).
Antibody Validation
All primary antibodies used in this study were validated in accordance with the recommendations of the International Working Group for Antibody Validation (IWGAV) (Uhlen et al. 2016) and the NIH guidelines for authentication of key biological resources (NOT-OD-17–068). For CREB2/ATF4 (mouse monoclonal clone B-3), specificity was confirmed by genetic knockdown (siRNA-mediated reduction of the target band), verification of the expected molecular weight in Western blotting, and consistency with previously published studies using the same clone. For p53, phospho-p53 (Ser15), and GADD45α antibodies, validation was supported by detection of single bands at the expected molecular weights and agreement with expression patterns reported in peer-reviewed studies under similar experimental conditions. RRIDs, catalog numbers, and manufacturers for all primary and secondary antibodies are provided in the corresponding sections.
Small Interfering RNA (siRNA) Transfection
The roles of GADD45α, CREB2, and p53 in glutamate-induced oxidative cytotoxicity were examined using mouse-specific siRNAs targeting GADD45α (sc-35439), CREB2/ATF4 (sc-35113), and p53 (sc-29436), along with a control siRNA (sc-37007) (Santa Cruz Biotechnology). HT22 cells were seeded to achieve 30–50% confluence at the time of transfection. Transfections were performed using 40 nM siRNA and Lipofectamine 2000 (Invitrogen, cat no. 11668019) following the manufacturer’s protocol. Cells were maintained for 48 h before analysis, and knockdown efficiency was confirmed by Western blotting.
RNA Isolation, cDNA Sythesis
Total RNA was extracted from glutamate-treated HT22 cells using TRIzol reagent (Invitrogen, cat. no. 15596026) according to the manufacturer’s protocol. RNA concentration and purity were assessed by spectrophotometry (A260/A280). For cDNA synthesis, 1 µg of total RNA was reverse-transcribed using M-MLV Reverse Transcriptase (200 U/µL; Thermo Fisher Scientific, cat. no. 28025013) and random hexamer primers (Roche Applied Science, cat. no. 11034731001). The 20-µL reverse transcription reaction contained 1× RT buffer, 0.5 mM dNTPs, 50 ng/µL random hexamers, 40 U RNase inhibitor (Thermo Fisher Scientific, cat. no. EO0381), and 200 U M-MLV reverse transcriptase. Reactions were performed at 25 °C for 5 min (primer annealing), 42 °C for 30 min (extension), and 95 °C for 5 min (enzyme inactivation).
Quantitative Real-Time PCR (qPCR)
Quantitative PCR was conducted using SYBR Green PCR Master Mix (Applied Biosystems, cat. no. 4367659) on an ABI PRISM 7300 real-time PCR system. The standard cycling conditions were as follows: 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. A melt-curve analysis (60–95 °C, 0.3–0.5 °C increments) was performed to verify amplification specificity. The primer sequences were: CREB2 (ATF4): forward 5′-GCCATTTCTGCTTGCTGTCT-3′; reverse 5′-ACCTAGGCTTTCTTCAGCCC-3′ GAPDH: forward 5′-GGCACAGTCAAGGCCGAGAA-3′; reverse 5′-CAGCAATGCATCCTGCACCA-3′ Relative gene expression levels were calculated using the 2^−ΔΔCt method (Fukui et al. 2009).
Measurement of Total and Lipid ROS in HT22 Cells
Total ROS: Intracellular ROS levels were measured using 2′,7′-dichlorofluorescein diacetate (H₂DCFDA; Molecular Probes, cat. no. D399). HT22 cells were treated with or without 5 mM glutamate for 8 h, then incubated with 10 µM H₂DCFDA in serum-free medium for 10 min at 37 °C in the dark. Cells were washed with HBSS. Total ROS Imaging (H₂DCFDA/DCF fluorescence): DCF fluorescence derived from H₂DCFDA oxidation was visualized using an AXIO fluorescence microscope (Carl Zeiss, Oberkochen, Germany) equipped with a FITC filter set (excitation 470–490 nm; emission 520–550 nm) and a Plan-Apochromat 20×/0.8 NA objective. Images were acquired at room temperature under identical exposure and gain settings for all samples. Fluorescence images were captured using a sCMOS camera at 16-bit depth, and representative fields were selected under non-saturating conditions to ensure accurate comparison of ROS levels.
Quantification of DCF Fluorescence Intensity: DCF fluorescence was quantified using ImageJ (NIH). All images were acquired under identical exposure and gain settings and analyzed as raw 16-bit files. Background was removed using the rolling-ball algorithm (50-pixel radius). Regions of interest (ROIs) were drawn around individual cells, and mean fluorescence intensity was measured using the “Measure” function. For each condition, 30–50 cells were quantified.
Lipid ROS Imaging (BODIPY™ 581/591 C11): Lipid peroxidation was assessed using BODIPY™ 581/591 C11 (Invitrogen, cat. no. D3861), which undergoes an oxidation-dependent emission shift from red (~ 590 nm) to green (~ 510 nm). HT22 cells grown on glass coverslips were treated as indicated, washed with HBSS, and incubated with 10 µM BODIPY C11 in HBSS for 20 min at 37 °C. After washing, coverslips were mounted and imaged using an LSM 900 confocal microscope (Carl Zeiss, Oberkochen, Germany) equipped with 488-nm and 561-nm laser lines and a Plan-Apochromat 40×/1.3 NA oil-immersion objective. Green (oxidized) and red (non-oxidized) fluorescence signals were acquired under identical exposure, gain, and detector settings across samples at 16-bit depth to ensure quantitative comparison of lipid peroxidation.
In-Vivo Animal Experiments
Mouse-derived BV2 microglial cells and HT22 hippocampal neuronal cells were utilized for mechanistic in vitro studies. For in-vivo validation, we employed a rat model, as the kainic acid (KA)–induced hippocampal injury model is particularly well-characterized in rats, offering high reproducibility of CA3-specific neuronal degeneration (Sperk 1994), whereas mice often exhibit strain-dependent variability or resistance to excitotoxic injury (McKhann et al. 2003). All procedures involving live animals were performed under an approved Institutional Animal Care and Use Committee (IACUC) protocol (protocol no. 2008 − 1767) at the University of Kansas Medical Center and adhered to NIH guidelines for the care and use of laboratory animals. Adult male Sprague–Dawley rats (250–270 g; Harlan, Indianapolis, IN, USA) were housed under a 12 h light/12 h dark cycle at controlled temperature (25 °C) and humidity (60%), with free access to food and water. Animals were allowed to acclimate for one week prior to experimentation and were randomly assigned to treatment groups (n = 5 per group).
Neurodegeneration induced by kainic acid (KA) was generated following the general methodology described by (He et al. 2006), with several modifications. Rats were anesthetized with ketamine/xylazine (50/5 mg/kg, s.c.) and placed in a stereotaxic frame. KA (1 µL of a 1 µg/µL solution) was bilaterally injected into the lateral ventricles using the following coordinates: anterior–posterior − 1.0 mm, medial–lateral ± 1.6 mm, dorsal–ventral 4.5 mm. Control rats received an equivalent volume of sterile saline. To prevent reflux, the injection needle was left in place for approximately 4 min before withdrawal, and the incision was then closed. At the end of the experimental period, rats were deeply anesthetized and perfused transcardially with 0.9% saline. Brains were removed, post-fixed overnight, cryoprotected in 30% sucrose, frozen, and coronally sectioned at 30 μm for histological, immunohistochemical, and Western blot analyses. Mouse-derived BV-2 microglial cells and HT22 hippocampal neuronal cells were used for in vitro mechanistic studies. For in-vivo experiments, rats were used, as the kainic acid–induced hippocampal injury model is well established in rats and allows reliable anatomical and subregional analysis.
Histological/Immunohistochemical Analysis
Histological evaluation included hematoxylin eosin (H&E) staining to assess overall cytoarchitecture and Fluoro-Jade B staining to detect degenerating neurons, following the method of Schmued and Hopkins (Schmued and Hopkins 2000) with minor modifications. Briefly, sections were treated with 0.06% potassium permanganate for 10 min, incubated in 0.01% Fluoro-Jade B for 20 min, rinsed, dried, and prepared for imaging.
For CREB2 immunohistochemistry, free-floating sections were incubated sequentially with mouse anti-CREB2 antibody (1:50; Santa Cruz Biotechnology), a biotinylated secondary antibody, and an avidin–biotin complex (Vector Laboratories, cat. no. PK-4000), followed by visualization with DAB. Sections were mounted, dehydrated, cleared, and coverslipped for microscopic analysis.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 10 (GraphPad Software, San Diego, CA). Sample sizes (n) were determined based on prior studies using the same experimental model and effect size, and the number of biological and technical replicates for each experiment is reported in the corresponding figure legends. Normality of data distribution was assessed using the Shapiro–Wilk test, Q-Q plot, and homogeneity of variance was evaluated using Brown-Forsythe. For normally distributed and variance-homogeneous data, one-way or two-way ANOVA followed by Bonferroni’s post hoc test was performed. Non-parametric tests were conducted when the data did not meet the assumptions. Exact p-values, test statistics (e.g., F(df1, df2), t(df)), and degrees of freedom are reported in APA style in each figure legend. All analyses were conducted with investigators blinded to group allocation.



Comments (0)