Cell culture
Human SH-SY5Y neuroblastoma cells were obtained from the Korean Cell Line Bank (Seoul, Korea). Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; HyClone, UT, USA) supplemented with 10% fetal bovine serum (FBS; HyClone) and 100 U/mL penicillin–streptomycin (HyClone). Cultures were grown at 37 °C in a humidified atmosphere containing 5% CO₂. To ensure experimental reliability, cell lines were routinely authenticated and screened for mycoplasma contamination before use.
HUWE1 overexpression and knockdown in SH-SY5Y cells
For overexpression experiments, SH-SY5Y cells were transfected with Myc-tagged HUWE1 wild-type (pCMV6-Myc-HUWE1 WT) or a C4341S mutant (pCMV6-Myc-HUWE1 C4341S), previously described by Lee et al. (Lee et al. 2024), using jetOPTIMUS® DNA transfection reagent (Polyplus-transfection, Illkirch, France) according to the manufacturer’s instructions. Cells were harvested 48 h post-transfection for protein analysis.
For gene knockdown, cells were transfected with synthetic HUWE1 siRNA (Genolution, Seoul, Korea) using Lipofectamine™ 2000 (Invitrogen, Carlsbad, USA) following the manufacturer’s protocol. Cells were collected 72 h after siRNA transfection for subsequent protein extraction. The HUWE1 siRNA sequence was identical to that previously reported (Lee et al. 2024).
Cell viability and cytotoxicity assays
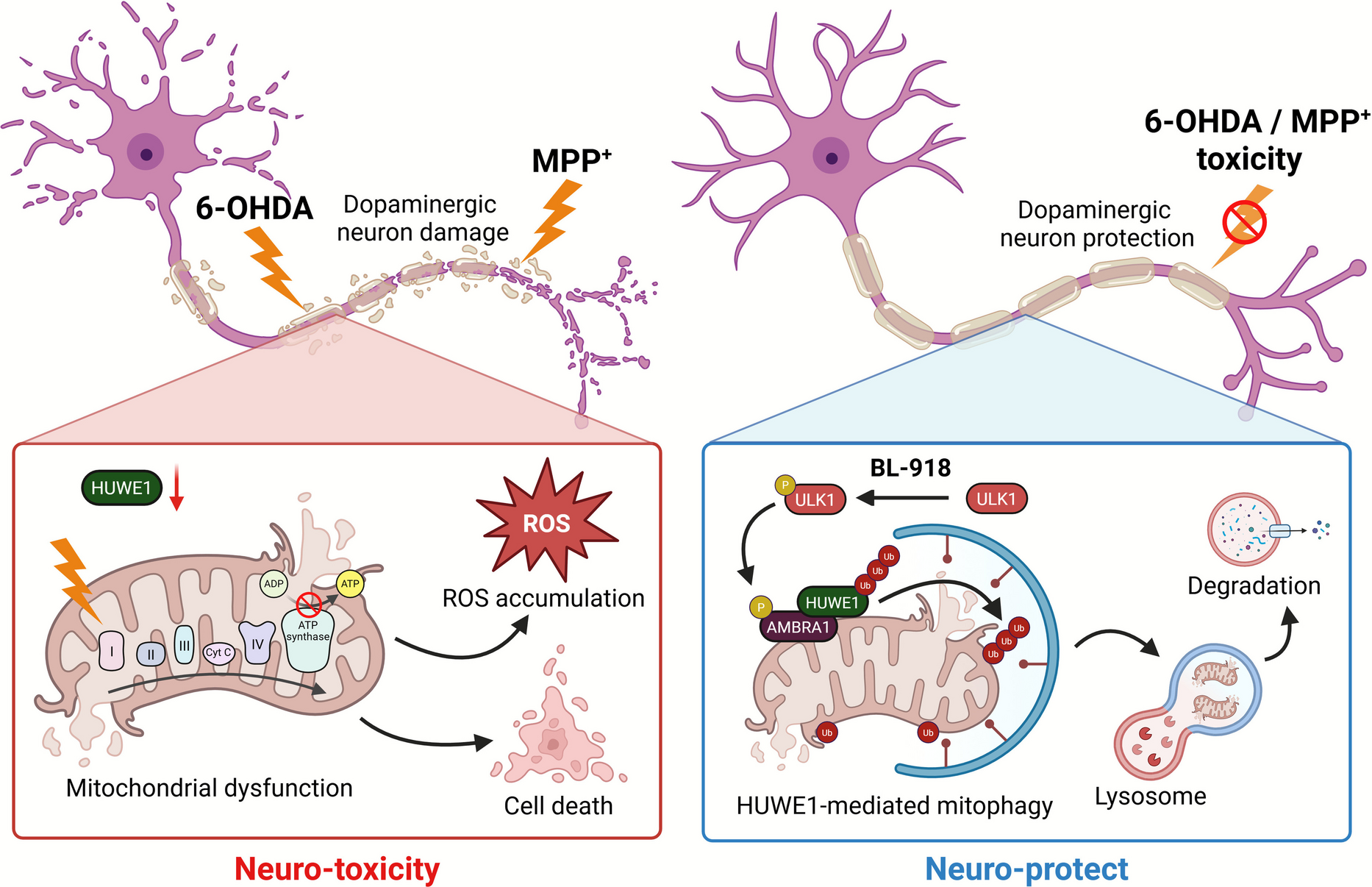
Cell viability and cytotoxicity were assessed using the Cell Counting Kit-8 (CCK-8; Dojindo, Kumamoto, Japan) and the LDH Cytotoxicity Detection Kit (Dojindo), respectively. SH-SY5Y cells were transfected with plasmids (24 h) or siRNA (48 h) and seeded at a density of 1 × 104 cells per well in 96-well plates. Cells were then exposed to 50 μM 6-hydroxydopamine (6-OHDA; Sigma-Aldrich, MO, USA) or 1 mM MPP⁺ (MedChemExpress, NJ, USA) in the presence or absence of 20 μM BL-918 (MedChemExpress) for 24 h. For the CCK-8 assay, reagent was added directly to the culture medium and incubated for 30 min at 37 °C, after which absorbance was measured at 450 nm using a microplate reader. For the LDH assay, culture supernatants were collected, and LDH activity was quantified following the manufacturer’s instructions, with absorbance recorded at 490 nm. Results were normalized to untreated controls.
Western blot analysis
Total cellular proteins were extracted using RIPA lysis buffer supplemented with protease inhibitors and lysed on ice for 30 min. Lysates were centrifuged at 13,000 rpm for 30 min at 4 °C, and the resulting supernatants were collected for protein quantification using the Bradford assay (Bio-Rad, CA, USA). Equal amounts of denatured proteins were separated by SDS-PAGE and transferred onto nitrocellulose membranes. The membranes were blocked with 5% non-fat milk for 1 h at room temperature and incubated overnight at 4 °C with the following primary antibodies: anti-HUWE1 (1:1,000, #9482, Cell Signaling Technology, MA, USA), anti-TOMM20 (1:1,000, #42,406, CST), anti-β-actin (1:1,000, sc-47778, Santa Cruz, CA, USA), anti-GAPDH (1:1,000, sc-32233, Santa Cruz), anti-Myc (1:1,000, #9B11, CST), anti-COX IV (1:1,000, #3E11, CST), anti-LC3B (1:1,000, #2775, CST), anti-Bcl-2 (1:1,000, sc-783, Santa Cruz), anti-Bax (1:1,000, sc-6236, Santa Cruz), anti-Cleaved caspase-3 (1:1,000, #9664, CST), anti-DRP1 (1:1,000, #8570, CST), anti-MFN2 (1:1,000, #9482, CST), anti-OPA1 (1:1,000, #80,471, CST), anti-p-ULK1 (1:1,000, #5869, CST), anti-ULK1 (1:1,000, A8529, ABclonal, MA, USA), anti-AMBRA1 (1:1,000, A1083, ABclonal), and anti-Ubiquitin (1:1,000, #39269, CST). After washing, the membranes were incubated with HRP-conjugated secondary antibodies (anti-mouse or anti-rabbit, 1:5,000, Santa Cruz) for 1 h at room temperature. Protein bands were visualized using an enhanced chemiluminescence (ECL) detection system (Biomax, Kyunggi-Do, Korea), and band intensities were quantified using ImageJ software.
Mitochondrial staining and immunofluorescence
SH-SY5Y cells were cultured on glass coverslips and stained with MitoTracker™ Red (Thermo Fisher Scientific, MA, USA) for 20 min at room temperature to label mitochondria. Cells were then fixed with 4% paraformaldehyde for 30 min, permeabilized with 0.1% Triton X-100 for 10 min, and blocked with 2% bovine serum albumin (BSA) in PBS. Cells were incubated overnight at 4 °C with primary antibodies against HUWE1 (1:1,000, #9482, Cell Signaling Technology, MA, USA), Myc (1:1,000, #9B11, CST), LC3B (1:1,000, #2775, CST), and AMBRA1 (1:1,000, A1083, ABclonal, MA, USA). After three washes with blocking buffer, cells were incubated for 1 h at room temperature with the corresponding Alexa Fluor-conjugated secondary antibodies (Alexa Fluor 488, 594, or 647). Nuclei were counterstained with DAPI, and fluorescence images were acquired using a confocal laser scanning microscope equipped with Airyscan (LSM980, Carl Zeiss, Oberkochen, Germany).
RNA extraction and qRT-PCR
Total RNA was extracted from SH-SY5Y cells using TRIzol™ reagent (Invitrogen, CA, USA) according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized from 2 μg of total RNA using GoScript™ Reverse Transcriptase (Promega, WI, USA). Quantitative real-time PCR (qRT-PCR) was performed using SYBR Green Real-Time PCR Master Mix (BIOFACT, Daejeon, Korea) on specific primers for each target gene. Primer sequences were as follows: HUWE1: F, GAGGAGTTGAAGCTACCTTGTG; R, CTGGTGTGATCCACTTTGG, SIRT1: F, TAGACACGCTGGAACAGGTTGC; R, CTCCTCGTACAGCTTCACAGTC, PPARGC1A: F, CCAAAGGATGCGCTCTCGTTCA; R, CGGTGTCTGTAGTGGCTTGACT, NFE2L2: F, CACATCCAGTCAGAAACCAGTGG; R, GGAATGTCTGCGCCAAAAGCTG, TFAM: F, GTGGTTTTCATCTGTCTTGGCAAG; R, TTCCCTCCAACGCTGGGCAATT, GAPDH: F, TCATCAGCAATGCCTCCTGC; R, GGCATGGACTGTGGTCATGA. Gene expression levels were normalized to GAPDH, and relative expression was calculated using the ΔΔCt method.
Mitochondrial fraction isolation
Mitochondrial fractions were isolated using a modified subcellular fractionation protocol. SH-SY5Y cells were washed with PBS and resuspended in mitochondrial fractionation buffer containing 10 mM Tris–HCl (pH 7.4), 250 mM sucrose, 20 mM HEPES (pH 7.4), 5 mM MgCl₂, 10 mM KCl, 1 mM EDTA, 1 mM EGTA, 0.02% digitonin, and protease inhibitors. Cells were homogenized by repeated aspiration through a 26-gauge needle attached to a syringe (10–20 passes). Homogenates were centrifuged at 900 × g for 2 min at 4 °C to remove nuclei and unbroken cells. The resulting supernatant was further centrifuged at 10,000 × g for 20 min at 4 °C, and the pellet was collected as the mitochondrial fraction.
Measurement of intracellular ROS
Intracellular reactive oxygen species (ROS) levels were measured using 2′,7′-dichlorofluorescin diacetate (DCFDA; Sigma-Aldrich, MO, USA). SH-SY5Y cells were seeded on glass coverslips and treated under experimental conditions. After treatment, cells were incubated with 5 μM DCFDA in serum-free medium for 20 min at 37 °C in the dark. Excess dye was removed by washing twice with PBS, and fluorescence images were acquired using a confocal laser scanning microscope equipped with Airyscan (LSM980, Carl Zeiss, Oberkochen, Germany) with excitation at 488 nm and emission collected at 525 nm. Fluorescence intensity was quantified using ImageJ software.
Mitochondrial functional analysis
Mitochondrial membrane potential (ΔΨm) and mitochondrial reactive oxygen species (MitoROS) were assessed using JC-1 (MedChemExpress, NJ, USA) and MitoSOX Red (MedChemExpress), respectively. SH-SY5Y cells were seeded on glass coverslips and treated under experimental conditions. For ΔΨm measurement, cells were incubated with 5 μM JC-1 in culture medium for 20 min at 37 °C in the dark, washed twice with PBS, and immediately imaged using a confocal microscope equipped with Airyscan (LSM980, Carl Zeiss, Oberkochen, Germany). JC-1 aggregates (red fluorescence, high ΔΨm) and monomers (green fluorescence, low ΔΨm) were detected at excitation/emission wavelengths of 561/590 nm and 488/530 nm, respectively. Mitochondrial membrane potential was quantified as the red-to-green fluorescence ratio using ImageJ software.
For MitoROS measurement, cells were incubated with 5 μM MitoSOX Red in serum-free medium for 20 min at 37 °C in the dark, washed twice with PBS, and fluorescence images were acquired using the same confocal microscope at excitation/emission of 510/580 nm. Fluorescence intensity was quantified using ImageJ software.
Measurement of cellular ATP levels
Cellular ATP content was determined using the ATP Determination Kit (Invitrogen, CA, USA) following the manufacturer’s instructions. SH-SY5Y cells were washed with PBS and lysed by boiling in sterile distilled water for 10 min. Lysates were centrifuged at 12,000 × g for 20 min at 4 °C to remove cellular debris, and the resulting supernatants were collected. ATP levels were quantified using a luminometer, and values were normalized to protein content or cell number as appropriate.
Mitochondrial electron transport chain enzyme activity assay
Activities of mitochondrial Complex I and Complex IV were measured using the Complex I Enzyme Activity Microplate Assay Kit (ab109721, Abcam, Cambridge, UK) and the Complex IV Human Enzyme Activity Microplate Assay Kit (ab109910, Abcam), respectively, according to the manufacturer’s instructions. Briefly, protein samples were captured by complex-specific antibodies pre-coated on microplate wells. Complex I activity was determined by spectrophotometric monitoring of NADH oxidation at 450 nm using 300 μg of protein per sample, while Complex IV activity was assessed by monitoring cytochrome c oxidation at 550 nm using 100 μg of protein per sample. Enzyme activities were calculated from the change in absorbance over time using the extinction coefficient specific to each assay.
Statistical analysis
All experiments were independently performed at least three times using biological replicates. Data are presented as mean ± standard error of the mean (SEM). Statistical comparisons between two groups were conducted using an unpaired Student’s t-test, with p-values less than 0.05 considered statistically significant. For comparisons involving two experimental groups, statistical significance was conducted using an unpaired Student’s t-test. For comparisons involving three or more experimental groups, one-way ANOVA followed by Tukey’s post hoc test was used, as the experiments were designed to compare distinct treatment conditions within a neurotoxin-induced stress paradigm rather than a fully crossed factorial design. All analyses were performed using GraphPad Prism software (version 6.00; GraphPad Software, CA, USA).

Comments (0)