Study design and treatment
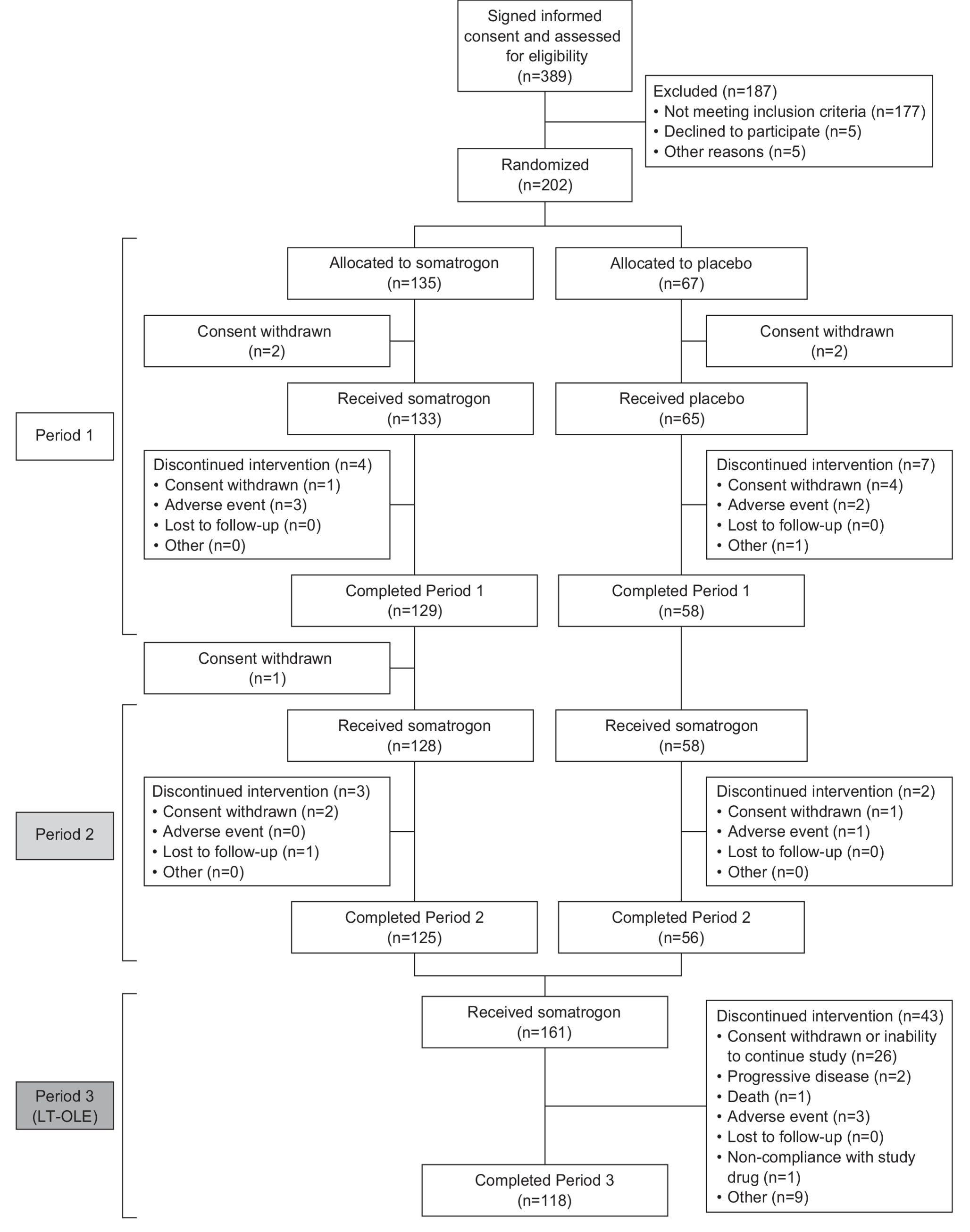
This phase 3 study (NCT01909479) was a randomized, double-blind, placebo-controlled, parallel-group, multicenter trial conducted in patients with aGHD. The study was sponsored by OPKO Biologics Ltd., a subsidiary of OPKO Health, Inc., and conducted between August 2013 and August 2018 at 71 sites in Australia, Austria, France, Georgia, Germany, Greece, Hungary, Israel, Poland, Romania, Russia, Serbia, Slovakia, South Korea, Spain, Taiwan, Ukraine, and the USA.
Patients were randomly assigned 2:1 (using an interactive response technology system) to receive either somatrogon or placebo. The starting dose of somatrogon/placebo was based on gender and age (Supplementary Table 1), as well as on whether female patients were taking oral estrogen therapy. The somatrogon dose was modified (Supplementary Table 2) as required to maintain IGF-I levels within the target range of − 0.5 ≤ IGF-I SDS ≤ + 1.5. To preserve blinding, dose modifications were also implemented for placebo recipients. All study treatments were self-administered as a once-weekly subcutaneous injection (using a needle and syringe) to the thighs or abdomen, with injections sites rotated. Patients received training on how to self-inject somatrogon/placebo, with the patient administering the first dose under the supervision of study site staff. Patients were provided with sufficient study drug to maintain dosing until the next scheduled study visit and were required to document their injections (injection site, dose, and vial number) in a patient diary that was regularly reviewed by study staff and at monitoring visits.
The study consisted of three treatment periods. Period 1 was a 26-week randomized, double-blind, placebo-controlled treatment period, and Period 2 was a 26-week open-label extension (OLE) followed by a 2- to 8-week washout period. During Period 2 all patients received somatrogon; patients originally randomized to placebo in Period 1 began with a starting dose of somatrogon based on their gender, age, and estrogen therapy, whereas patients originally randomized to somatrogon continued on the same dose as they had completed during Period 1. Period 3 was designed as a multiyear long-term OLE (LT-OLE) study to evaluate long-term safety. During Period 3, patients continued receiving the same dose of somatrogon received at the end of Period 2. For clarity, throughout this manuscript we refer to patients who were originally randomized to somatrogon in Period 1 as the somatrogon group and those who were randomized to placebo in Period 1 as the placebo group.
The primary objective of the study was to demonstrate the clinical superiority of somatrogon over placebo in terms of decreasing trunk fat mass (FM, in kg) in patients with aGHD; this objective was defined, in part, according to findings from a clinical study described by Hoffman et al. [23]. Secondary objectives included assessing the efficacy of somatrogon (vs. placebo) for other body composition parameters and evaluating the safety and tolerability of somatrogon compared with placebo. Additional post hocsupplemental efficacy analyses were also undertaken following discussions with regulatory agencies (see Supplementary Methods for rationale).
This study was approved by the Institutional Review Board or Independent Ethics Committee of the participating institutions and followed the Declaration of Helsinki and International Council on Harmonisation Good Clinical Practice guidelines. Signed and dated informed consent was obtained from each patient prior to any participation in study-related procedures.
Patients
Adult men and women (≥ 23 to ≤ 70 years of age) with GHD were eligible for inclusion in this study. Patients using hormonal replacement therapy for other hypothalamic-pituitary axis deficiencies could be included provided they had been on an optimized and stable treatment regimen for at least 3 months prior to screening. At screening, patients had to have IGF-I levels ≤ − 1 SDS, adjusted for age and gender [24]. Patients had to have a body mass index (BMI) between 23.0 and 35.0 kg/m2 (inclusive), with no GH therapy for at least 9 months prior to the study. Patients also had to be on a stable diet and exercise regime and were not to have plans to modify their diet or exercise for ≥ 12 months at the time of enrollment. Key exclusion criteria were overt history of diabetes mellitus, evidence of growth of a benign intracranial tumor within the last 12 months, suspected or diagnosed ongoing cancer or history of any cancer, and women who were pregnant or breast-feeding. Other typical inclusion/exclusion criteria are shown in the Supplementary Methods. While participating in the study, patients were informed to avoid making changes to their diet, exercise regime, or smoking habits. However, diet and exercise were not regularly monitored during the study.
Study assessmentsEfficacy
The primary efficacy endpoint was change in trunk FM (in kg) from Baseline to Week 26, measured using dual-energy X-ray absorptiometry (DEXA). Secondary efficacy endpoints included changes (from Baseline to Week 26 and Week 52) in total FM, LBM, and percentage change in trunk FM. Change in trunk FM from Baseline to Week 52 was also a secondary endpoint. The following efficacy endpoints were evaluated using DEXA at screening, Week 15, and Week 26 in Period 1; Week 39 in Period 2; and at the end of Period 2 (Week 52/end-of-study): trunk FM, total FM, LBM, and bone density (only at screening and end-of-study OLE). IGF-I levels were measured centrally at screening, baseline and at day 3 or 4 post dose at Weeks 3, 7, 11, 15, 19, 23, 26, 32, 39, 45, and 52 by chemiluminescence IGF-I immunoassay [24]. The schedule of assessments conducted at each visit is shown in Supplementary Fig. 1.
Two post hoc supplemental efficacy analyses were conducted. The first analysis was performed to assess the impact of outliers (rationale and details provided below), and the second one evaluated additional body composition endpoints, including change in percent trunk FM (defined as 100 times trunk FM divided by the sum of trunk FM and trunk LBM), change in trunk LBM, and change in appendicular skeletal muscle mass (ASMM).
Safety
Safety evaluations included all adverse events (AEs), electrocardiogram, fundoscopy, and laboratory assessments (consisting of glucose metabolism, IGF-I levels, and immunogenicity). Treatment-emergent AEs (TEAEs) were defined as any AE that occurred on or after initiation of treatment. All AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA 19.1). The intensity or severity of an AE was characterized as mild, moderate, or severe. Local injection site reactions were recorded as AEs, and injection site pain scores of 8 or higher were recorded as an AE. Anti-drug antibodies (ADAs) were assessed from serum samples using qualitative, validated methods [16].
Statistical analyses
The safety analysis set consisted of all randomized patients who received at least one dose of somatrogon or placebo. The efficacy analysis set consisted of all patients in the safety population who had a primary efficacy evaluation at Week 26. The efficacy endpoints and changes from Baseline were summarized by descriptive statistics for each treatment group (see Supplementary Methods for details of the statistical comparisons). Safety endpoints were summarized using descriptive statistics.
Following database lock and unblinding of the data, unusually high total body mass and change in trunk FM were noted for six patients. As a result, a post hoc sensitivity analysis was conducted to assess the impact of outliers on the results of the study. These post hoc sensitivity analyses consisted of (i) identification of outliers, (ii) tipping-point analysis, and (iii) non-parametric approaches (see Supplementary Materials for further details).



Comments (0)