
Remember me




The yeasts used in this work were isolated from soil samples from Unidade de Ensino, Pesquisa e Extensão em Produção de Grandes Culturas e Bioenergia (UEPE-GCBE Aeroporto), belonging to the Universidade Federal de Viçosa (UFV), Viçosa, Minas Gerais, Brazil. Yeasts were maintained in Yeast extract–peptone (YP) medium [% (w/v): 0.50 yeast extract, 0.50 peptone] with 30% (v/v) glycerol at -80 ºC (Kurtzman et al. 2011). The isolates are stored in the culture collection of the Laboratory of Microbial Physiology (LABFIS) of the Department of Microbiology from UFV. Before each experiment, yeasts were activated by transferring 0.20 mL stock into a 125 mL Erlenmeyer flask containing 30 mL of Yeast extract–peptone-dextrose (YPD) medium [% (w/v): 0.50 yeast extract, 0.50 peptone and 1.00 glucose] and incubated at 30 ºC and 200 rpm for 18 h (G25, New Brunswick, USA), as commonly used for yeast cultivation (Smith and Burke 2014). The suspension was centrifuged at 12,000 g for 10 min (Sigma D-37520, Osterode am Harz, Germany), and the cells were washed with 0.85% (w/v) NaCl. Before inoculation, the optical density at 600 nm (OD600), measured using a UV–visible spectrophotometer (BECKMAN DU series 600, Beckman Instruments Inc., USA), was adjusted to reach 0.1 in the beginning of each cultivation.
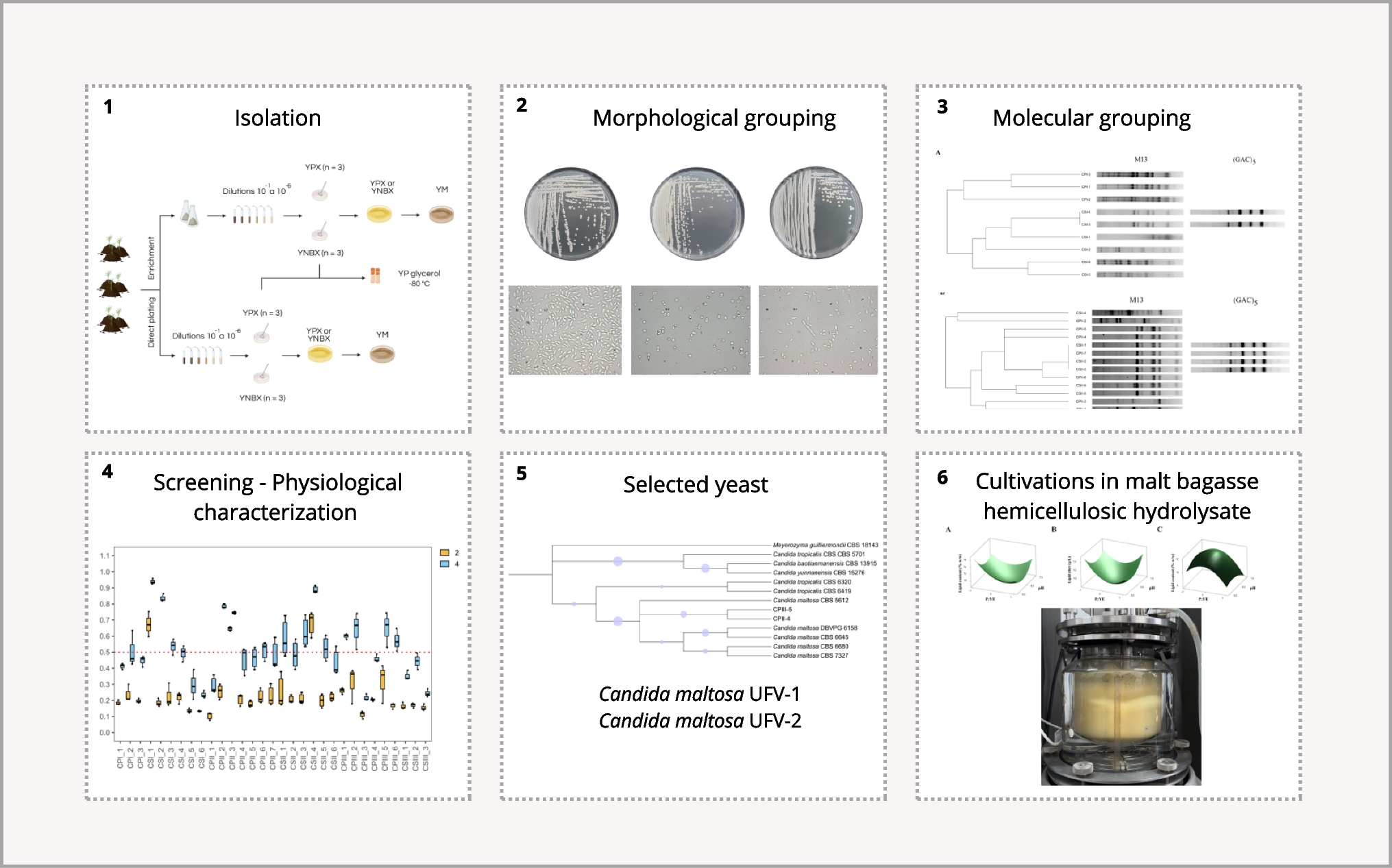
Soil sampling and yeast isolationSamples were collected from UEPE-GCBE Aeroporto (-20.748964, -2.842144), a station focused on agricultural research and the production of large crops and bioenergy. Composite samples were collected from three different points in three different collection areas, both at the surface (depth of 0 cm) and at a depth of 10 cm, with three portions taken at each point to ensure greater representation of soil conditions throughout the area analyzed.
Isolation was performed as described by Yurkov et al. 2016 and Kurtzman et al. 2011, with modifications, employing two strategies: direct plating and enrichment culture of environmental samples. For direct plating, one gram of composite soil sample was suspended in 9 mL of sterile 0.85% (w/v) NaCl solution (10⁻1 dilution) in a 15 mL tube and homogenized by vortex for 2 min. Serial dilutions (10⁻1 to 10⁻6) were prepared in sterile 0.85% (w/v) NaCl solution, and 100 µL aliquots were spread-plated onto solid Yeast extract–peptone-xylose (YPX) [% (w/v): 0.50 yeast extract, 0.50 peptone, 1.00 xylose] and Yeast nitrogen base-xylose (YNBX) [% (w/v): 0.67 yeast nitrogen base, 1.00 xylose] medium, both supplemented with 0.01% (w/v) chloramphenicol and 1.75% (w/v) agar. Spread plating was performed to ensure even distribution of cells. For the enrichment strategy, one gram of soil sample was inoculated into 50 mL of liquid YPX or YNBX medium in 125 mL Erlenmeyer flasks and incubated at 30 ºC, 200 rpm for 24 h. The culture was serially diluted (10⁻1 to 10⁻6) in sterile 0.85% (w/v) NaCl solution and plated onto their respective solid media (YPX-agar and YNBX-agar) using the same plating technique as for direct samples. The plates were incubated at 30 ºC and monitored daily during a 21-day incubation period, with macroscopic evaluations of colony morphology and microscopic evaluations of yeast cell structure to ensure accurate selection and purification of the isolates.
Morphological characterization and groupingThe morphotypes of yeast cells and colonies were analyzed with at least one representative of each morphotype being purified and preserved for later identification. The purified colonies were cultivated on Yeast extract–malt extract–xylose (YMX) medium [% (w/v): 0.30 yeast extract, 0.50 peptone, 0.30 malt extract, 1.00 xylose] with 1.75% (w/v) agar, incubated at 30 °C for 48 h. Plates were then photographed to evaluate macroscopic characteristics (Canon EOS Rebel T7, EF-S 18–55 mm), and slides were prepared for microscopic analysis (BX50F4, 40x, Olympus Optical Co. Ltd.), following the procedure described by Kurtzman et al., (2011).
PCR-fingerprintingFirst, the genomic DNA of yeast isolates was extracted from pure cultures using phenol–chloroform-isoamyl alcohol method adapted from Bartlett and Stirling, (2003). Briefly, the isolates were first activated, cell lysis was performed by cryogenic disruption using liquid nitrogen, followed by mechanical agitation with glass beads in extraction buffer [50 mmol/L Tris–HCl, 150 mmol/L NaCl, 20 mmol/L EDTA, 10 mmol/L HCl, and 0.20% (w/v) SDS] for 3 min using a vortex. Proteins were precipitated with phenol:chloroform:isoamyl alcohol (25:24:1) and subsequently centrifuged for 10 min at 12,000 g. The resulting material was treated with 10 mg/mL RNAse (Sigma) and then precipitated in sodium acetate (3 M, pH 5.2) and 100% (v/v) ethanol overnight at -20 ºC. DNA samples were centrifuged and washed with 70% (v/v) ethanol. The DNA was rehydrated with 30 µL of deionized water at 60 °C for 1 h and stored at -20 °C.
PCR-fingerprinting analyses were performed with primers M13 (5′-GAGGGTGGCGGTTCT-3′) and (GAC)5 (5’-GAC GAC GAC GAC GAC-3’) (Andrade et al. 2006; Baleiras Couto et al. 1996). Primer M13 was initially used to generate banding patterns for all isolates. When isolates from the same location produced identical profiles, indicating the presence of genetically similar lineages, the complementary primer (GAC)5 was applied to achieve further discrimination between the isolates or to indicate that they represented the same original strain. Each PCR assay was carried out in a total volume of 25 μL containing: 1.0 μL input DNA, 1.0 μL of 10 mM dNTP (Promega), 4.0 μL of 25 mM MgCl2 (Promega), 1.5 μL of 10 μM primer (IDT), 1.0 μL 5 U Taq DNA Polymerase (Promega), 4.0 μL 5X PCR buffer (Promega) and sterile deionized water. PCR assays were performed in the PCR Express thermal cycler (Bio-Rad) as follows: initial denaturation at 94 °C for 2 min, followed by 40 cycles of 45 s of denaturation at 95 °C, 1 min of primer annealing at 50 °C and 1 min of extension at 72 °C, and a final extension for 6 min at 72 °C. PCR products were separated by 1.5% (w/v) agarose gel electrophoresis in 1.0X TBE buffer (1.11 M Tris-base, 0.1 M boric acid, 0.5 M EDTA, pH 8) for 180 min at 60 V, and visualized under UV light after staining with 0.05% (v/v) ethidium bromide (Sigma Aldrich, Milan, Italy) in a gel photodocumentation system (L-Pix). For band size comparison across gels, a control DNA sample from our isolates was included in every agarose gel run, allowing direct alignment and normalization of band patterns between different samples. The electrophoretic bands for each sample were hand marked and transformed into a binary matrix, which was then used for hierarchical grouping and represented as dendrograms using the “dendextend” R package (Galili 2015). Clustering and image analysis were conducted in R (v. 4.2.1).
Screening of yeast isolatesFirst screeningYeasts were activated and the cell suspensions were inoculated into 96-well microplates containing 200 µL of 0.67% (w/v) YNB medium supplemented with 2% (w/v) glucose or xylose in triplicate. Growth was monitored using the OD600 over 24 h for glucose and 48 h for xylose using a Multiskan GO microplate reader (ThermoScientific, Wilmington, DE, USA). Isolates that reached OD600 ≥ 0.5 in xylose were selected for further analysis. Growth in the presence of 2.0 g/L acetic acid was further assessed for the selected strains (triplicates). Isolates reaching OD600 ≥ 0.5 at the highest acetic acid concentration were selected for oleaginous phenotype screening.
Second screeningThe yeasts selected in the first screening step were evaluated for their potential to produce lipids. The yeasts were inoculated into 125 mL Erlenmeyer flasks containing SS2 medium (Tanimura et al. 2014) 100:1 (C:N) [% (w/v): 0.010 yeast extract, 0.052 (NH4)2SO4, NaCl 0.010, CaCl2 0.010, MgSO4 0.050] supplemented with 3% (w/v) xylose. This medium was used to promote lipid accumulation under nitrogen limitation, a condition known to trigger lipid biosynthesis in oleaginous yeasts (Chattopadhyay and Maiti 2021). In this formulation, (NH₄)₂SO₄ was the main nitrogen source used to define the C:N ratio, while yeast extract was maintained at a low fixed concentration solely as a source of growth factors. The experiment was performed in duplicates and cultivations were carried out for 96 h at 30 ºC and 200 rpm. Isolates presenting the oleaginous phenotype [lipid content ≥ 20% (w/w)] were selected and subsequently tested in the same medium supplemented with 1.0 g/L acetic acid, under the same conditions to assess its impact on lipid production. Biomass and lipid production were determined at the end of each cultivation. Cell dry weight (DW) was determined after freeze-drying and lipid quantification was conducted using the SPV (Sulfo-Phospho-Vanilin) method as described by Knight et al., (1972) with modifications. Both methods are detailed in Section "Analytical methods".
D1/D2 sequencingThe best isolates selected in the second screening were subjected to taxonomic identification after differential grouping in the fingerprinting step initially by sequencing the D1/D2 domains of the gene encoding the 26S subunit of ribosomal DNA. The universal primers NL-1 (5′-GCATATCAATAAGCGGGAGGAAAAG-3′) and NL-4 (5′-GGTCCGTGTTTCAAGACGG-3′) were used for amplification of the D1/D2 region (Lachance et al. 1999). PCR was performed in a final volume of 50 μL, containing: 2.0 μL DNA, 1.0 μL of each primer (10 μmol) (IDT), 5.0 μL 5X delivery (Promega), 2.0 μL 25 mM MgCl₂ (Invitrogen), 2.0 μL 10 mM dNTP (Promega), 0.2 μL Taq DNA polymerase (5U) (Promega), and sterile deionized water. PCR was performed in a PCR Express thermocycler (Bio-Rad) with the following program: initial denaturation at 95 °C for 2 min, followed by 5 cycles of denaturation at 95 °C for 15 s, annealing at 54 °C for 25 s, and extension at 72 °C for 20 s, with a final extension at 72 °C for 10 min. PCR products were analyzed by 1.5% (w/v) agarose gel electrophoresis (Sigma Aldrich, Milan, Italy) in 1.0X TBE, for approximately 120 min at 80 V. Gels were stained with ethidium bromide (Sigma Aldrich, Milan, Italy), visualized under ultraviolet light, and photographed in a gel documentation system (L-Pix).
The PCR products were sequenced by Sanger method at BPI Biotechnology (São Paulo, Brasil). The obtained sequences were processed for taxonomic identification using the Geneious software (Version 2025.0.3, Dotmatics, Auckland, New Zealand) and the BLAST tool at NCBI (National Center for Biotechnology Information). After sequencing, raw files were imported into Geneious by creating a new folder and uploading the files corresponding to the forward and reverse reads. The sequences were aligned and assembled into contigs, with low-quality regions automatically trimmed during this process. Once the contigs were assembled, they were annotated by conducting a BLAST search against the NCBI nr database. For each sample, the best taxonomic match was identified based on the lowest e-value and higher sequence identity. The phylogenetic positioning of the isolates selected as potential lipid producers was represented by a tree constructed from the sequences of the D1/D2 region. The sequences used were obtained from different sources according to The Yeasts Database (https://theyeasts.org/species-search) and selected based on the highest genetic similarity. The sequences were aligned using the ClustalW algorithm (Thompson et al. 1994). The phylogenetic tree was constructed using the Maximum Likelihood method with the Kimura 2-parameter model, selected as the most appropriate evolutionary model based on log-likelihood. The bootstrap consensus tree was inferred from 1,000 replicates; branches reproduced in fewer than 50% of replicates were collapsed. The dataset included 13 nucleotide sequences with 570 positions in the final alignment. The rate variation model allowed for a proportion of evolutionary invariable sites. Meyerozyma guilliermondii CBS 18143 was used as the outgroup to root the tree. Phylogenetic analysis was conducted in MEGA version 12 (Kumar et al. 2024) and the resulting tree was edited and visualized in the Interactive Tree Of Life (iTOL) platform (Letunic and Bork 2021).
Impact of different carbon/nitrogen ratios on the oleaginous phenotypeThe most promising oleaginous yeasts, that is, those that presented an oleaginous phenotype even in the presence of acetic acid in the previous screening step, were evaluated regarding lipid production under different carbon/nitrogen (C:N) ratios. The strains were inoculated into 125 mL Erlenmeyer flasks containing SS2 medium with varying C:N ratios (50:1, 75:1, 100:1, 125:1, and 150:1). Nitrogen concentrations were adjusted using (NH₄)₂SO₄ to final values of 0.109, 0.071, 0.052, and 0.041% (w/v), respectively, with a fixed concentration of xylose [3% (w/v)]. Cultivation was carried out at 30 °C and 200 rpm for 72 h in triplicates. At the end of cultivation, biomass and lipid production were determined as described in Section "Analytical methods". Results were analyzed using one-way ANOVA followed by Tukey’s post-hoc test to identify significant differences between treatments (p < 0.05).
Growth in the presence of furfural, hydroxymethylfurfural and formic acidThe same strains assessed regarding the oleaginous phenotype in different C:N ratios were inoculated into 96-well microplates containing 200 µL of either 0.67% (w/v) YNB or YP medium (0.5% yeast extract, 0.5% peptone) with 2.0% (w/v) xylose. The media were supplemented with increasing concentrations of furfural (0.05; 0.10; 0.50; 1.00; 2.00 g/L), hydroxymethylfurfural (HMF) (0.10; 0.50; 1.00; 2.00; 4.00 g/L), and formic acid (0.20; 0.50; 1.00; 1.50; 2.00 g/L). The concentrations were selected to represent values found in lignocellulosic hydrolysates (Kumar et al. 2020; Vanmarcke et al. 2021). To assess whether the solvent used to dilute furfural had any effect on yeast growth, ethanol was added to the culture medium at the same concentration present in the highest furfural treatment (1.65 g/L, corresponding to furfural at 2.00 g/L). Cultivations were performed in quadruplicate at 30 ºC for 96 h. The OD600 was measured every 24 h using a Multiskan GO microplate reader (ThermoScientific, Wilmington, Delaware, USA).
Preparation of malt bagasse hemicellulosic hydrolysateThe hemicellulosic hydrolysate obtained from malt bagasse was used to evaluate its applicability as a substrate for lipid production. The pretreatment was conducted by the Laboratory of Bioprocesses and Sustainable Products, Escola de Engenharia de Lorena, Universidade de São Paulo (USP). The biomass was initially air-dried, milled using a knife mill (Benedetti 270, Benedetti, Brazil), and sieved to reach an average particle size of 4.7 mm. Acid pretreatment was carried out using diluted H₂SO₄ (Vetec®) in 200 mL stainless steel reactors operating as a closed system and maintained in a thermostatic water bath. The pretreatment conditions were 2% (v/v) sulfuric acid (H2SO4, Vetec®), 165 °C, and 40 min of reaction. After pretreatment, the hydrolysates were cooled, filtered, and stored at 4 °C until further use Mesquita et al. 2016). Proteins and other particles in suspension were removed from the hydrolysate before quantifying sugar and inhibitors, following the precipitation protocol of Siegfried et al. (1984). Glucose, xylose, arabinose, acetic acid, formic acid, furfural, and HMF were determined by High-Performance Liquid Chromatography (HPLC) performed by the authors, using a RID-20A refractive index detector (Shimadzu, Japan) and an Aminex HPX-87H column (300 × 7.8 mm) (Bio-Rad, USA) at 45 °C, with 5 mM sulfuric acid as the mobile phase and a flow rate of 0.7 mL/min.
Preparation of detoxified and non-detoxified hemicellulosic hydrolysateThe hydrolysate was prepared in both detoxified and non-detoxified forms to evaluate their effects on microbial growth and lipid production (Section "Effect of pH and nitrogen source on yeast growth and lipid production in malt bagasse hemicellulosic hydrolysate" and "Batch bioreactor cultivation of C. maltosa UFV-1 using detoxified malt bagasse hemicellulosic hydrolysate"). For the preparation of non-detoxified hydrolysate, the pH was first adjusted to the target cultivation (see Section "Effect of pH and nitrogen source on yeast growth and lipid production in malt bagasse hemicellulosic hydrolysate") value using sodium hydroxide (NaOH) beads under constant stirring, followed by centrifugation at 4000 g for 10 min. The supernatant was vacuum filtered using Whatman filter paper, supplemented with nitrogen sources (see 2.11), then filtered using a 0.22 µm membrane and stored at 4 °C until use.
The hydrolysate detoxification process was carried out as described by Marton et al. 2006, with adaptations. The detoxification process began with pH adjustment to 7.0 using calcium oxide (CaO) under continuous agitation, followed by centrifugation at 3000 g for 15 min. The supernatant was then acidified to pH 2.5 with phosphoric acid (H₃PO₄) and treated with activated charcoal 1% (w/v) at 60 °C and 150 rpm for 30 min. After vacuum filtration through a Whatman paper, the hydrolysate underwent the final preparation steps: readjustment of the pH to the target culture value (see Section "Effect of pH and nitrogen source on yeast growth and lipid production in malt bagasse hemicellulosic hydrolysate" and "Batch bioreactor cultivation of C. maltosa UFV-1 using detoxified malt bagasse hemicellulosic hydrolysate") using NaOH beads, centrifugation at 4000 g for 10 min, supplementation with nitrogen sources (see Section "Effect of pH and nitrogen source on yeast growth and lipid production in malt bagasse hemicellulosic hydrolysate" and "Batch bioreactor cultivation of C. maltosa UFV-1 using detoxified malt bagasse hemicellulosic hydrolysate"), filtration through a 0.22 µm membrane, and storage at 4 °C.
Effect of pH and nitrogen source on yeast growth and lipid production in malt bagasse hemicellulosic hydrolysateTo investigate the influence of pH and nitrogen source on yeast growth and lipid production in malt bagasse hemicellulosic hydrolysate, the following experimental strategy was employed: (i) preliminary evaluation of the effects of pH and nitrogen source in non-detoxified hydrolysate; (ii) optimization step performed through a Central Composite Rotational Design (CCRD) targeting growth and lipid production in non-detoxified hydrolysate; and (iii) validation of the conditions predicted by the model using non-detoxified and detoxified media. The optimization of pH and nitrogen source was prioritized due to the intrinsic characteristics of the hemicellulosic malt bagasse hydrolysate obtained by acid hydrolysis, which typically presents very low pH (often < 1) and limited availability of assimilable nitrogen. These factors directly affect yeast growth and lipid accumulation through their influence on inhibitor toxicity and C:N balance (Beopoulos et al. 2009). In this stage, peptone and yeast extract were used as nutritional supplements to support growth in the complex hydrolysate matrix.
In the preliminary evaluation, yeasts were cultivated in 96-well microplates, incubated at 30 ºC for 50 h under agitation using a Multiskan GO microplate reader (ThermoScientific, Wilmington, DE, USA). Cultivations were carried out in non-detoxified hydrolysate to evaluate the effects of pH and nitrogen source on cell growth with pH values adjusted to 2.0, 3.0, 4.0, 5.0, 6.0 and 7.0 using NaOH, and two nitrogen sources (peptone or yeast extract, both at 1.0 g/L). These experiments were conducted with four biological replicates.
Based on the results obtained in the microplate cultivations, a Central Composite Rotational Design (CCRD) with response surface methodology (RSM) was applied to determine the optimal initial pH and peptone:yeast extract ratio – P:YE, with growth and lipid production as response variables. The experiment comprised 11 samples for each yeast, with 3 replicates at the central point (Table 1). The independent variables were evaluated at five levels (− α, − 1, 0, + 1, + α), allowing the estimation of linear, quadratic, and interaction effects. Cultivations were conducted in 250 mL Erlenmeyer flasks containing 75 mL of medium and incubated at 30 °C and 200 rpm for 60 h. The dependent variables analyzed during optimization were specific growth rate (µ; h-1), final biomass (g/L), lipid content [% (w/w)] and lipid titer (g/L). Data was first analyzed using an ANOVA to evaluate the significance of the main effects and interactions. A predictive mathematical model was fitted to the data based on a second-order polynomial equation, according to Eq. 1.
$$y=_+\sum _+_+\sum _+_^}+\sum _+__$$
(1)
where y represents the response variable, that is, growth rate (µ; h-1), final biomass (g/L), lipid content [% (w/w)] or lipid titer (g/L); \(_\) and \(_\) are the independent variables (pH and the P:YE), and \(_\),\(_\), \(_\) and \(_\) are the estimated model coefficients. The quality of the fit was evaluated using the coefficient of determination (R2) and the significance of the model terms. Response surface plots and contour graphs were generated to visualize the optimal cultivation regions.
Table 1 Experimental range and levels of independent variables used in the Central Composite Rotational Design (CCRD)After defining the optimal cultivation conditions, the predicted model was validated using detoxified and non-detoxified hydrolysates, applying the same parameters used during the optimization step (30 °C, 200 rpm, 75 mL) for 60 h. Samples were collected at the beginning and end of cultivation and subjected to a protein precipitation protocol described by Siegfried et al. (1984) prior to HPLC analysis. Glucose, xylose, arabinose, acetic acid, formic acid, furfural, HMF, glycerol, and ethanol were then quantified as described in Section "Preparation of malt bagasse hemicellulosic hydrolysate". At the end of cultivation, biomass was also collected for lipid quantification and fatty acid profile characterization.
Batch bioreactor cultivation of C. maltosa UFV-1 using detoxified malt bagasse hemicellulosic hydrolysateThe cultivations were carried out in duplicate in 1.3 L bench-top bioreactors (BioFlo/CelliGen 115, Eppendorf, Germany) containing 900 mL of detoxified hemicellulosic malt bagasse hydrolysate supplemented with 1 g/L peptone and 200 mg/L chloramphenicol. The addition of the antibiotic aimed to prevent bacterial growth arising from residual spores that might persist in the hydrolysate after the filtration process. Operational conditions were maintained at 400 rpm, with the addition of Antifoam 204 (Sigma-Aldrich) at a final concentration of 400 µL/L. Unlike the other experiments, the cultivation temperature was set to 31 °C, since preliminary assays indicated higher growth rate and biomass accumulation by C. maltosa UFV-1 under this condition. The initial volumetric oxygen transfer coefficient (kLa), previously determined by the nitrogen gassing-out method (Garcia-Ochoa and Gomez 2010) based on dissolved oxygen concentration measurements, was 93.03 h⁻1. No cascade control strategy was employed to maintain the dissolved oxygen concentration. The initial optical density (OD₆₀₀) was adjusted to 0.2, and the total cultivation time was 48 h. At the end of cultivation, the biomass was harvested and used for lipid quantification.
Analytical methodsSpecific growth rateSpecific growth rate (µ; h−1) was calculated by applying a linear regression to the ln (OD600) readings plotted against time (h) during the exponential growth phase of cell growth.
Dry biomassAt the end of cultivation, 40 mL of the medium was centrifuged at 4,000 g for 10 min. The pellet was washed twice with a 0.85% (w/v) NaCl solution. After washing, the biomass was freeze-dried for 24 h to ensure complete dehydration. The dry biomass was then determined gravimetrically, and the concentration calculated in g/L based on the initial volume of the culture medium.
Lipid quantificationLipids were quantified using the SPV method as described by Knight et al., (1972) with adaptations. The phosphor-vanillin (PV) reagent was freshly prepared by dissolving 600 mg of vanillin into 10 mL of absolute ethanol, subsequently diluted to 500 mL with 90 mL of water and 400 mL of 85% (w/v) phosphoric acid. For the standard curve, commercial soybean oil was diluted in methanol:chloroform 2:1 to a final concentration of 1.5 mg/mL; the curve comprised a lipid mass ranging from 50–250 µg. For quantification, cell dry biomass (10 mg) was diluted in 2 mL of water. Then, the 100 µL of the biomass dilution was mixed with concentrated sulfuric acid (2 mL) and incubated at 100 °C for 10 min. After cooling to room temperature (20 °C), 2 mL of the PV reagent was added for the colorimetric reaction. The mixture was incubated at 37 °C for 15 min, then transferred to 96-well flat-bottom microplates for absorbance measurements. Absorbance was measured at 530 nm using a Multiskan FC plate reader (Thermo Scientific, San Jose, CA, USA).
Fatty acid profileFatty acid profile analysis was performed according to the Sherlock Microbial Identification System protocol (version 6.0, Microbial ID, Inc., Newark, DE, USA). For fatty acid extraction, 4–5 mg of freeze-dried biomass was subjected to saponification and methylation steps to produce fatty acid methyl esters (FAMEs). FAMEs were extracted and analyzed using a gas chromatograph (Agilent 7890A, Agilent Technologies, Santa Clara, CA, USA) equipped with a fused silica capillary column coated with 5% phenylmethyl silicone and a flame ionization detector (FID), following the default configuration of the Sherlock system. Fatty acids were identified by comparison with the RTSBA6 library using Sherlock MIDI software.
HPLC analysisThe concentration of xylose (0.75 to 30.0 g/L), glucose (0.75 to 30.0 g/L), arabinose (0.15 to 6.00 g/L), acetic acid (0.05 to 6.00 g/L), formic acid (0.01 to 6.00 g/L), furfural (0.02 to 0.80 g/L), HMF (0.02 to 0.80 g/L), glycerol (0.13 to 5.00 g/L) and ethanol (0.13 to 5.00 g/L) was determined by HPLC as follows: LC-20AT HPLC system (Shimadzu, Japan) coupled to a RID-20A refractive index detector (Shimadzu, Japan) and an Aminex HPX-87H ion exchange column (300 × 7.8 mm, 9 μm, Bio-Rad, Munich, Germany). The mobile phase used was 5 mM H2SO4 with a flow rate of 0.7 mL/min at 45 °C.
Determination of lipid production parametersThe lipid production parameters were determined as follows:
$$Lipid content (\% w/w) = (P/DW)\times 100$$
(2)
where \(P\) is final lipids (mg) (see 2.2.11.3); DW is cell dry weight (mg).
$$Lipid titer (g/L)=Lipid content (\% w/w) \times _$$
(3)
where \(_\) is final biomass (g/L) (see 2.2.11.2).
$$Lipid Productivity (g/L h)=\frac$$
(4)
where t is the total cultivation time (h).
$$Lipid Yield (_)=\frac_-_}$$
(5)
where \(_\) ≡ final sugar concentration (g/L); \(_\) ≡ initial sugar concentration (g/L). The sugar concentration includes the sum of glucose, xylose and arabinose in cultivations with hemicellulosic hydrolysates and pure sugars in defined media during the screening steps.
Comments (0)