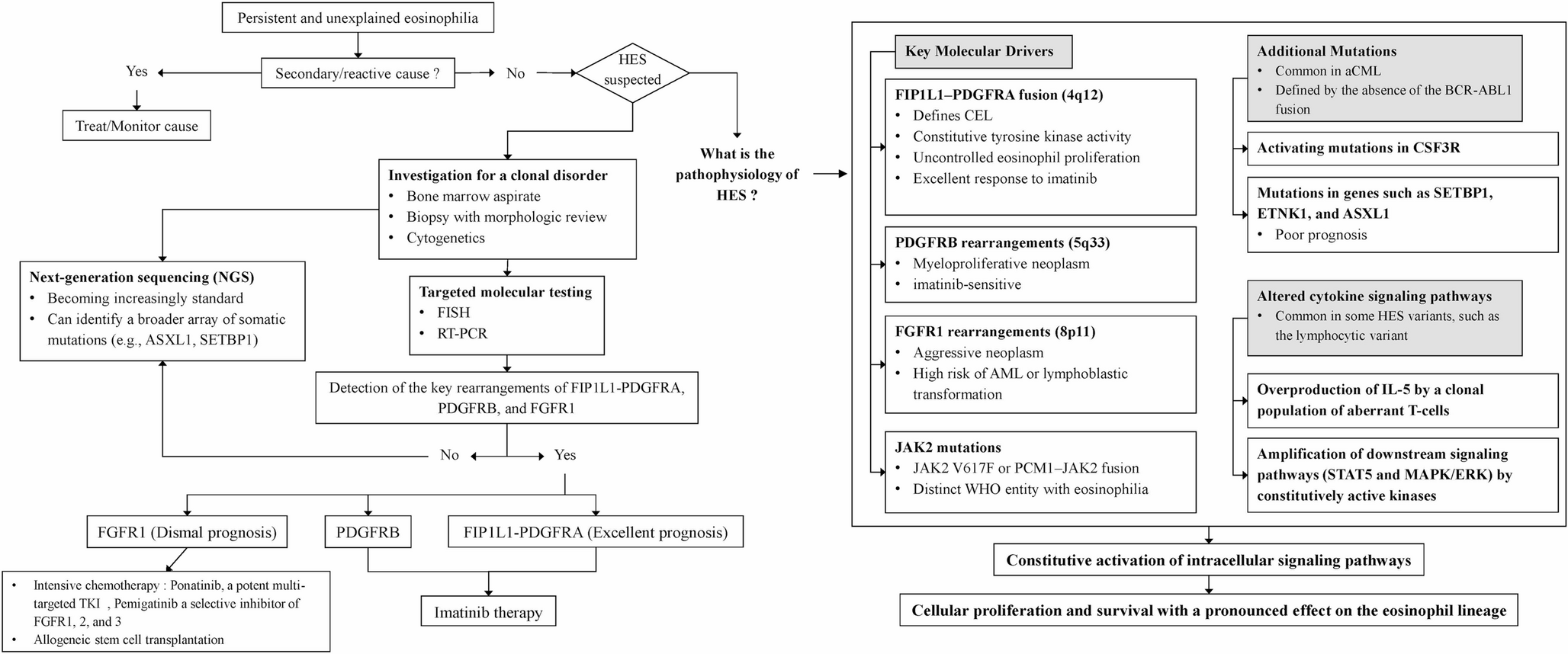
Several practical conclusions emerge from the literature. First, BRAF-mutated CLL is uncommon but real, and the mutation spectrum is fundamentally different from what clinicians may expect from hairy cell leukemia or melanoma. In other hematologic malignancies, BRAF often behaves as a more canonical driver lesion: BRAF V600E is a defining hallmark of hairy cell leukemia and a major MAPK lesion in Langerhans cell histiocytosis and related histiocytic neoplasms [2, 3]. CLL is different. The predominance of non-V600E lesions is one of the most reproducible findings across broad sequencing studies and should shape both interpretation and therapeutic speculation [12,13,14, 16, 18].
Cytogenetics and Co-Mutations
At the cytogenetic level, trisomy 12 emerged as the most repeatedly reported chromosomal abnormality co-occurring with BRAF mutations. This finding is consistent with prior observations that RAS–BRAF–MAPK pathway lesions are enriched in trisomy 12 CLL, supporting the concept that BRAF-mutated CLL may preferentially cluster within a distinct biologic subgroup. By contrast, del(11q), del(17p), and del(13q) were reported less often and appeared more unevenly across studies [10,11,12,13, 15].
Another important observation from Fig. 1 is the marked between-study heterogeneity in both the number and pattern of co-alterations. Some studies reported only a limited number of recurrent partners, whereas others demonstrated broader co-mutational complexity. This likely reflects both biologic diversity and methodological differences. In particular, studies using broader next-generation sequencing panels or whole-exome approaches tended to identify a wider range of co-alterations than earlier targeted studies. Likewise, cohorts composed of relapsed, refractory, or progression-phase patients appeared more likely to show multiple concurrent abnormalities than treatment-naïve cohorts, consistent with clonal evolution under therapeutic pressure [4, 6, 7, 9, 14].
Taken together, these findings suggest that BRAF-mutated CLL is best understood as part of a broader cooperative genomic framework, rather than as a stand-alone lesion. The recurrent co-occurrence with TP53, NOTCH1, SF3B1, and trisomy 12 supports the view that BRAF mutations may mark a biologically distinct subset characterized by genomic complexity, signaling activation, and potentially more aggressive disease behavior. However, the currently available evidence remains largely study-level and descriptive. Therefore, these co-alteration patterns should be interpreted as hypothesis-generating rather than definitive evidence of a uniform molecular signature [10, 12, 13].
Prognostic Implications and Clinical Impact
The prognostic significance of BRAF alterations in CLL remains uncertain. Only a small number of studies directly compared outcomes between BRAF-mutated and BRAF-wild-type cases, and in most of these studies the number of BRAF-mutated patients was very limited. As a result, robust comparative estimates were generally not available, and most outcome data were descriptive [10, 12, 14, 15].
Overall, the available evidence does not support BRAF as an established independent prognostic marker in CLL. Instead, BRAF alterations seem to occur more often in biologically higher-risk settings, including trisomy 12-enriched disease, IGH-associated genomic subgroups, MAPK-pathway–activated disease, and therapy-exposed relapse. When clinical associations were reported, they more often involved earlier need for treatment or shorter treatment-free intervals than a consistent adverse effect on overall survival. This suggests that treatment-timing endpoints, such as time to first treatment, treatment-free survival, or time to next treatment, may better reflect the clinical relevance of BRAF than overall survival, although these outcomes were not uniformly reported across studies [4,5,6, 9, 10, 12, 13, 13, 14, 14, 15, 20].
Taken together, the most balanced interpretation is that BRAF is not currently suitable for routine risk stratification in CLL. Rather, it is better viewed as a marker of adverse biological context in selected subsets. Its main clinical value may lie in interpretation alongside co-mutation profile, cytogenetic background, and disease phase, particularly when detected at relapse or progression on targeted therapy [4, 6, 9, 10, 12, 13, 13, 14, 14, 15].
BRAF in Treatment-Exposed Disease and Resistance
Studies from the targeted-therapy era suggest that the significance of BRAF in CLL lies less in baseline prevalence and more in its role at relapse. BRAF alterations have been reported in patients progressing on BTK inhibitors, PI3K inhibitors, and venetoclax-based regimens, often together with other signaling abnormalities rather than as isolated lesions [4,5,6, 9, 14]. This pattern supports the idea that BRAF participates in clonal evolution and pathway-level escape under treatment pressure, rather than acting as a single dominant resistance mechanism.
Murali et al. provided important mechanistic support for this concept by showing that MAPK-pathway activation can mediate resistance to PI3K inhibition in relapsed CLL [9]. Likewise, Bonfiglio et al. and Brown et al. identified BRAF alterations in patients progressing on BTK inhibitors, including some cases without detectable BTK or PLCG2 mutations [4, 6]. Herling et al. described a non-V600E BRAF lesion during venetoclax resistance evolution [14], while Jain et al. showed that relapse after fixed-duration ibrutinib-venetoclax may occur even in the absence of canonical BTK, BCL2, or PLCG2 mutations [5]. Together, these observations suggest that relapse in CLL cannot always be explained by the usual resistance mutations alone and that MAPK-pathway activation may represent an alternative route of disease escape in a subset of patients.
In the venetoclax setting, the most appropriate conclusion is therefore not that BRAF-mutated CLL is uniformly resistant to BCL2-directed therapy, but that MAPK-pathway lesions may contribute to relapse biology in selected cases. This also provides a biologic rationale for dual-targeted strategies, such as BTK inhibitor plus BCL2 inhibitor combinations, which may better suppress parallel survival pathways and reduce dependence on any single escape route. However, this remains a pathway-level, hypothesis-generating interpretation rather than evidence for a BRAF-specific treatment recommendation [5, 14].
Richter Transformation and Why this Review Matters
A possible association between BRAF and Richter transformation has been reported, although the evidence remains limited. The strongest support comes from a small pathology-based study in which BRAF V600E was identified more often in Richter syndrome than in untransformed CLL [16]. This finding is important because it contrasts with the broader CLL literature, where non-V600E variants predominate. As a result, V600E-positive Richter-transformed disease should not be interpreted as representative of BRAF-mutated CLL as a whole.
This distinction also clarifies the clinical relevance of the review. The importance of BRAF in CLL lies less in its overall frequency and more in whether it identifies biologically meaningful subsets at important clinical transitions, including earlier treatment need, targeted-therapy relapse, and transformation. For this reason, BRAF remains relevant to contemporary precision-based interpretation of CLL, even if it does not yet have a routine role in frontline risk stratification [4,5,6, 10, 12, 14, 16].
What is Known About BRAF-Directed Therapy?
Direct evidence for BRAF inhibitors in CLL is very limited, and the currently available data do not support routine extrapolation from melanoma or hairy cell leukemia. Jebaraj et al. reported limited evidence of cell-death induction with BRAF inhibition in primary CLL cells [18]. More detailed functional work by Giménez et al. showed that vemurafenib did not meaningfully inhibit ERK phosphorylation in primary MAPK-pathway–mutated CLL cells, while dabrafenib showed only modest and partly nonspecific effects; in contrast, the downstream ERK inhibitor ulixertinib more effectively suppressed phospho-ERK in mutated samples [12].
These findings fit the underlying biology: most CLL-associated BRAF lesions are non-V600E and are embedded within broader signaling networks. Moreover, paradoxical ERK activation with BRAF inhibition has been described in CLL-relevant settings, reinforcing caution about indiscriminate use of single-agent BRAF inhibitors [21]. The one context in which BRAF-directed therapy may be more plausible is transformed, V600E-positive Richter syndrome, but even there the evidence remains limited to small case-based or pathology-based observations rather than prospective CLL trials [12, 16, 21].

Comments (0)