
Remember me
Clonal hematopoiesis (CH) arises when hematopoietic stem cells acquire somatic mutations that confer a proliferative advantage, leading to the expansion of mutant clones. Initially associated with the aging process, CH is understood to be influenced by inherited genetic factors. Recent genome-wide association studies of CH carriers have identified multiple genetic loci where germline variations influence predisposition to CH [1, 2]. Specifically, germline variations in DNA damage repair (DDR) genes have emerged as key determinants of CH risk later in life.
The DDR genes collectively preserve genomic integrity by activating checkpoints, coordinating repair mechanisms, and inducing cellular apoptosis or senescence in the event of unsuccessful repair. Proper functioning of DDR genes is crucial for hematopoietic stem cells as they experience continual genotoxic stress from DNA replication, environmental exposure, and cancer therapies such as radiation and cytotoxic chemotherapy. Therefore, it is unsurprising that germline variations in DDR genes may create a permissive environment for the acquisition and survival of somatic mutations that underlie CH. Mutations in DDR genes are also frequently observed in various hematologic disorders. In this review, we examine the impact of germline DDR gene variants on CH and myeloid neoplasms.
Germline Predisposition to Clonal HematopoiesisClassic hereditary hematologic disorders are often characterized by pathogenic germline mutations that directly contribute to the syndromic pathophysiology and follow Mendelian patterns of inheritance. The age at presentation of such disorders varies with patients developing symptoms in childhood (e.g. Noonan syndrome, Fanconi Anemia, Tatton-Brown-Rahman), or in adulthood (e.g. ANKRD26, ETV6, RUNX1, CEBPa) [3, 4]. Widespread use of next generation sequencing (NGS) used primarily to detect somatic changes that occur with malignancy and often portend risk [5,6,7] or response to therapy [8,9,10] has uncovered genetic changes that occur at 45–55% allele burden (thus implying possible germline variants). Most commonly, this occurs among patients with previously deemed idiopathic hematologic malignancies. The implication of these germline variants has been further complicated by variable penetrance patterns and complex interactions with other genes. For example, while germline DDX41 mutations are inherited in an autosomal dominant fashion, only 25–50% of DDX41 carriers develop DDX41-associated acute myeloid leukemia at a median age of onset of 71 years [11, 12]. And while many germline variants are sufficient to lead to a disease phenotype, some exert their influence and disease predisposition in how they impact the acquisition of somatic changes.
Conceptually, this was first commonly recognized in familial clusters of myeloproliferative neoplasms (MPN) patients with somatic JAK2-V617F variants [13, 14]. Importantly, a shared germline haplotype GGCC “46/1” predispose a 3–4 fold risk of developing a somatic JAK2 -V617F variant which is causative of MPN [14,15,16]. Subsequently, genome-wide association studies (GWAS) of patients with JAK2 -V617F-CH and MPN revealed additional associations with germline variants in TERT, SH2B2, TET2, ATM, CHEK2, TINT and GFI1B genes [17]. Of these, different non-coding TERT variants have been shown to confer variable risks for MPN development, with intron 2 variants having significant association with more aggressive MPN phenotype and intron 3 variants associated with more indolent course of asymptomatic CH [17, 18].
Mosaic loss of the Y chromosome (mLOY), the most common somatic genetic lesion in men is associated with clear germline predisposition [18,19,20]. Initially mLOY was found to be associated with common SNP near the 5’ end of TCL1A gene (rs2887399), and subsequent large scale GWAS studies aiming to identify functional transcripts that contribute to mLOY identified eight additional contributing genes, many of which are involved in cell cycle progression and DNA damage response [19,20,21].
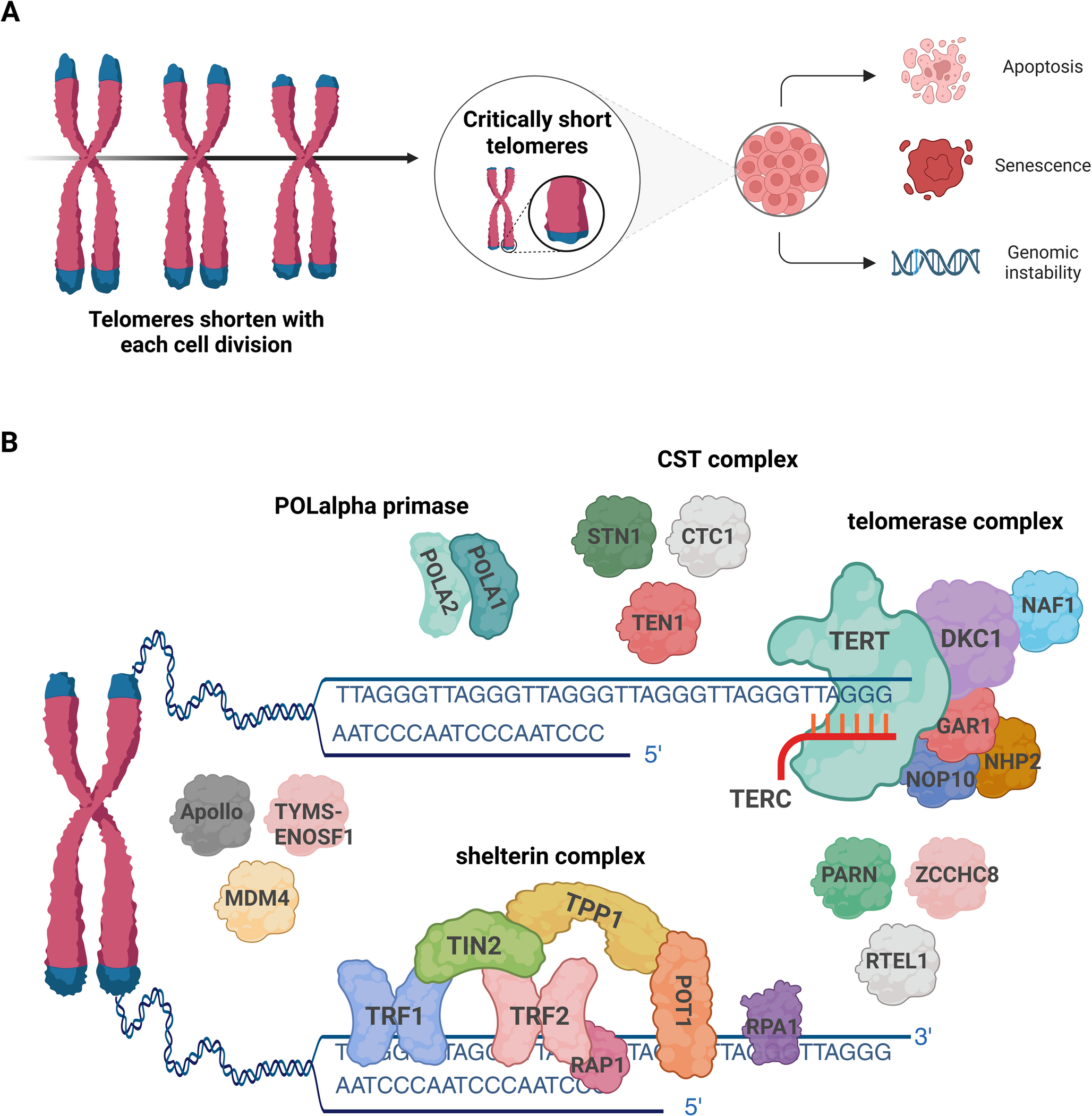
Mutations in DNA Damage Repair Genes and Predisposition to Clonal HematopoiesisCarcinogenesis is marked by an accumulation of mutations that provide a clonal advantage and ultimately genomic instability. This is a feed-forward process with increasing genomic instability leading to oncogene activation or loss of a tumor suppressor function [22, 23]. Genes involved in DNA Damage Repair (DDR) pathways are essential to monitoring and coordinating DNA repair, safeguarding the cells from propagating tumorigenic mutations during cellular division. Complex signaling cascades involved in DDR are mediated by checkpoint kinases. Mutations within DDR pathways, including ATM, CHEK2, BRCA1/2, PALPB2 and TP53 have been associated with development of a broad spectrum of tumors [24, 25]. While somatic mutations in DDR genes contribute to neoplastogenesis, particularly in selection of resistant cells after therapy, the germline influence of germline variants in DDR have impact across tumor types (Fig. 1).
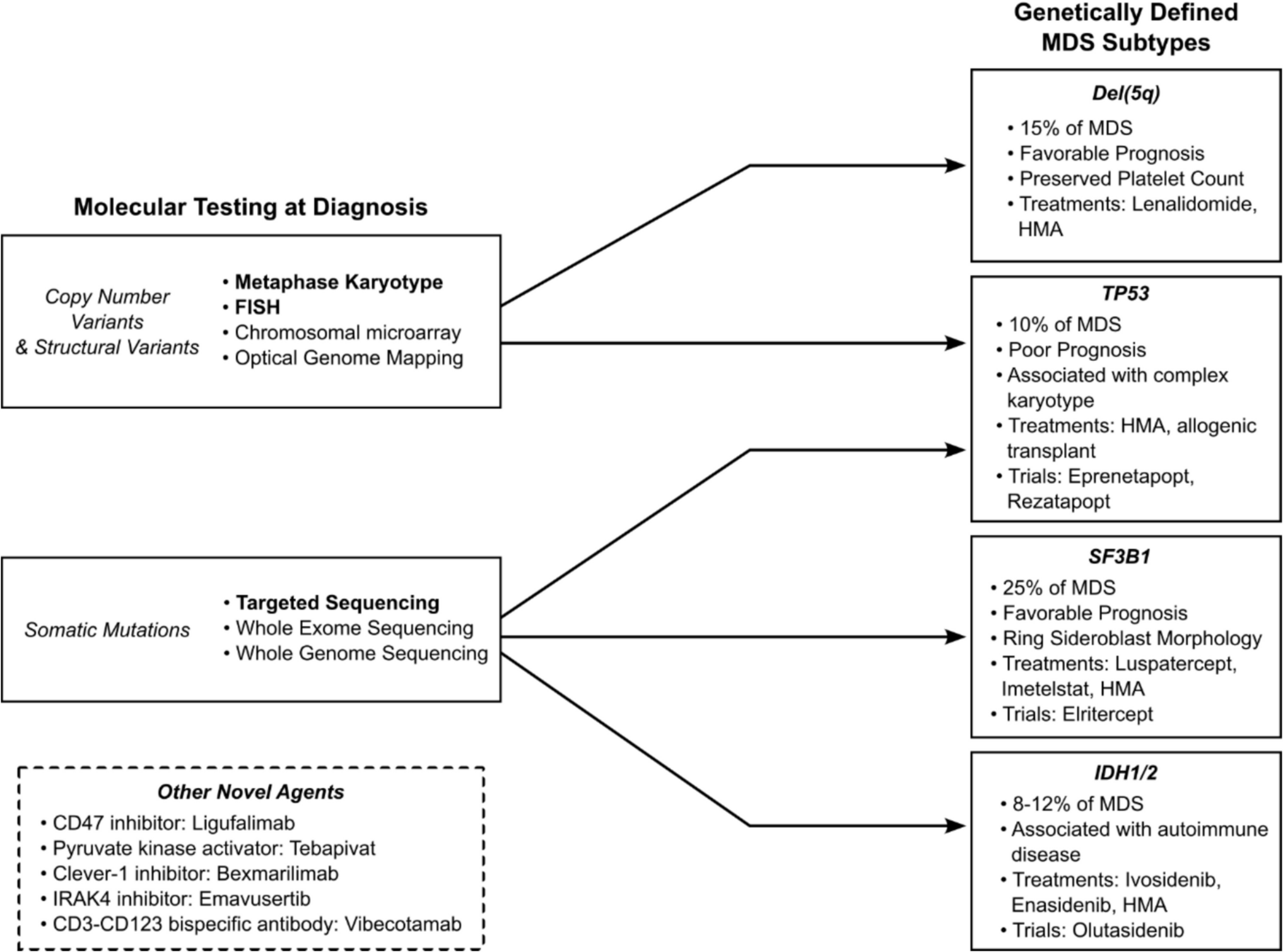
Fig. 1 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.ATM signalling in DNA damage repair (DDR response)
ATM is the nexus of DNA damage response. After extrinsic DNA damage, activated ATM phosphorylates several downstream targets including CHK2 and BRCA1 (not visualized). CHK2 is essential to the propagation of DDR signal to the downstream effectors including but not limited to p53 and BRCA1. Activated p53 engages p21 to trigger cell cycle arrest to allow for DNA repair, or BAX/BIM/PUMA cascade to initiate apoptosis. p53 also recruits a negative regulator PPM1D which functions to terminate DDR response once DNA repair is complete. Following activation by CHK2, BRCA1 recruits BRCA via PALB2 which then activates RAD51 for homologous recombination repair
Sustained efforts to characterize and understand factors contributing to CH have revealed a link between germline mutations in DNA damage repair genes and the development of CH. A large scale genome-wide association study (GWAS) on 184,121 people of European ancestry compared 10,203 individuals with CH to 173,918 without CH and identified 7 independent genome wide loci, including genes in DDR pathway, associated with risk of development of CH [1]. Here we discuss the important established links between germline DDR mutations and CH (Table 1).
Table 1 Summary of Germline DDR mutations associated with malignancies and impact on development of clonal hematopoiesisGermline ATM MutationsAtaxia telangiectasia mutated (ATM) is a principal DNA damage checkpoint serine/threonine kinase central to DNA double-strand break repair, particularly during G1-S and G2-M checkpoints of the cell cycle [26, 27]. The most well recognized condition associated with germline ATM mutations is ataxia-telangiectasia, an autosomal recessive neurodegenerative disorder characterized by progressive cerebellar ataxia, cutaneous telangiectasias, immunodeficiency, increased risk of lymphoid malignancies, and radiosensitivity [27, 28]. The onset of malignancies associated with ATM deficiency appear to stem from the mutation burden of ATM mutations. Homozygous ATM mutations uniquely predispose to development of childhood lymphoid malignancies due to ATM dependent repair of DNA double-stranded breaks during V(D)J recombination and class switch in B and T lymphocyte development [29, 30]. By contrast, heterozygous pathogenic ATM variants are associated with increased risks of malignancy in adulthood [31, 32].
Although ATM mutations drive hematologic malignancies, ATM mutations occur in less than 1% patients with AML and MDS [33]. The mechanisms via which ATM contributes to myeloid leukemogenesis remain poorly defined but are thought to stem from impaired cell cycle checkpoints ultimately leading to accumulation of genetic lesions and chromosomal instability, consistent with ATM’s critical role in DDR [20, 34].
While ATM mutations are not considered to be the founding events of myeloid leukemogenesis they have been associated with impaired self-renewal of hematopoietic stem and progenitor cells (HSPCs) and have been implicated in providing selective advantage to mutated HSPC [35]. Consequently, expansion of such mutated HSPCs could contribute to the development of clonal hematopoiesis. Recent genome-wide and exome-wide association analyses of healthy volunteer samples from United Kingdom (UK) Biobank and Geisinger Health System (GHS) identified rare germline ATM variants that were significantly associated with CH [2]. Separate analysis of UKB data also implicated ATM as gene associated with CH [1]. Additionally, germline ATM has been linked to increased risk of JAK2 V167F-CH in healthy population and to JAK2 V617F mutated MPN in previously diagnosed patients. This risk was independent from that of previously known predisposition emerging form 46/1 JAK2 haplotype [17].
Prospective tumor-blood paired sequencing of 46,906 patients from MSK-IMACT initiative, primarily including patients with advanced Stage III-IV cancers, showed that germline ATM variants were specifically associated with clonal hematopoiesis. Additionally, there was a strong association between germline ATM and somatic ATM-CH, with similar findings observed in patients with germline TP53 mutations, suggesting that bi-allelic inactivation may provide growth advantage, select for larger clonal size and thus contribute to clonal expansion [36]. Separately, single-nucleotide polymorphism associated with ATM has also been identified as one of causative genes and predisposition to mLOY which in its turn predisposes to further clonal hematopoiesis [19, 20].
Germline CHEK2 MutationsCHEK2 gene encodes for the checkpoint kinase 2 (CHK2), an effector serine/threonine kinase with critical role in cell cycle regulation, functioning as a tumor suppressor [37]. CHEK2 variants were first described among families with Li- Fraumeni-like syndrome without germline TP53 mutation [38]. Truncating variant c.1100delC is the most studied CHEK2 variant and carries a strong association with increased risk of early-onset breast cancer (37% cumulative risk). The association with other cancers such as gastric, colorectal, kidney, prostate, sarcoma and thyroid is less well established [37, 38].
Mutations in germline CHEK2 are seen in 2–3% of general population with the incidence higher in certain populations (p.S428F in Ashkenazi Jewish population and p.I200T in Eastern European populations). The majority of CHEK2 mutations are germline with somatic variants being quite rare [39]. Mutations in CHEK2 were previously thought to be uncommon in hematologic malignancies, with more recent studies suggesting that the true incidence may be as high as 15%, with certain populations having higher frequencies [39,40,41,42]. In large public AML/MDS databases such as BEAT AML, Leucegene, AML PMP, CHEK2 mutations were found in 1.2% of AML patients [41]. The incidence was higher in patients suspected to have hereditary hematologic malignancy syndrome, where 3.6% were found to have CHEK2 variants, with majority having p.I157T (53%), followed by pT367fs (c.100delC, 11%) variants [41].
Surprisingly, whole exome sequencing on paired leukemia and skin biopsy samples revealed 13.6% leukemia patients had germline variants in CHEK2. These findings were replicated in a small study of post-remission bone marrow samples of 47 patients with AML who were not suspected to have hereditary predisposition [43, 44]. Among 205 germline variants detected across 110 genes, only 11 were classified as pathogenic or likely pathogenic, with most localizing to DNA damage response genes, including ATM, CHEK2, FANCA, FANCM [43].
Germline CHEK2 mutations have been linked to development of CH in multiple large scale GWAS [1, 2, 17, 36]. In analysis of UKB and GHS data, germline CHEK2 truncating variants were strongly associated with DNMT3A-CH [2]. A separate analysis of a smaller dataset from the UK Biobank revealed that the c.100delC germline CHEK2 variant confers a markedly increased risk of DNMT3A-CH with the effect size 4 times larger than that of the other common risk alleles such as ATM, TERT, or SCM4 [1]. In gene-based association tests for aggregation of rare germline variants among participants of the National Heart, Lung, and Blood Institute Trans-omics for Precision Medicine (TOPMed) cohort while no genes reached exome-wide significance, CHEK2 was found to be the top associated gene [45]. Similar to germline ATM, CHEK2 has been closely associated with JAK2-V617F-CH and MPN [17].
Collectively, these findings underscore the importance of CHK2 in preserving genomic stability in hematopoietic stem cells and provide a mechanistic link between inherited DNA repair defects and the development of clonal hematopoiesis with high risk for subsequent myeloid malignancy.
Germline Mutations in TP53TP53 gene, commonly referred to as the “Guardian of the genome” encodes p53 tumor suppressor protein. P53 is essential to regulating cell cycle arrest, apoptosis, and cellular senescence in response to in response to cellular stress signals, particularly DNA damage [46]. Li-Fraumeni syndrome – a rare autosomal dominant syndrome defined by germline mutations in TP53 – confers a strong predisposition to broad spectrum of malignancies including breast cancer, soft tissue sarcomas, osteosarcomas, brain tumors, adrenocortical carcinomas, leukemia, and lymphoma [47, 48]. Hematologic malignancies, lymphoid as well as myeloid, in Li-Fraumeni syndrome are less common, affecting less than 10% of carriers [49,50,51,52].
In a recent study of Li-Fraumeni syndrome associated hematologic malignancies, age at the time of diagnosis ranged significantly, with B-ALL developing at a median age of 12 years old, non-Hodgkin lymphoma at 39 years old, and MDS/AML at 33 years old [49]. The patients diagnosed with hematologic malignancies without prior chemotherapy exposure were younger (14 years) and had predominantly lymphoid phenotype (81% ALL), compared to those with prior chemotherapy exposure who were older (31 years) and had myeloid phenotype (79% MDS/AML). Nearly half of those diagnosed with secondary myeloid malignancies had multi-hit TP53 loss.
Li-Fraumeni syndrome is phenotypically and genomically diverse, ranging from individuals with personal and family history of multiple neoplasms fulfilling clinical criteria despite the absence of a germline TP53 mutation, to asymptomatic germline TP53 carriers lacking any personal or family history of malignancy [47].
The diagnostic challenge is further compounded by the risk of misclassifying high-allele frequency TP53 mutations identified by sequencing of the peripheral blood samples as germline. Several studies of paired blood-tumor samples highlight the need for more nuanced diagnostic approach than peripheral blood testing as corresponding tumor samples may not contain any TP53 alterations and thus are not part of Li-Fraumeni syndrome spectrum [53, 54].
The analysis of paired blood-tumor samples from MSK-IMPACT revealed that of 17,992 patients with solid malignancies only 50 (0.28%) individuals had TP53 mutations, with only 38 (0.19%) confirmed to be of germline origin. Yet still, confirmed germline TP53 does not directly correlate with full Li-Fraumeni phenotype. Loss of heterozygosity (LOH) is the critical molecular event that leads to tumorigenesis that manifests as Li-Fraumeni syndrome. While 96% of the patients with true Li-Fraumeni tumor phenotype having concurrent LOH of wild-type TP53 and germline TP53 mutation, only 46% of non-Li-Fraumeni tumor phenotype patients have such double-hit TP53 loss [55].
Furthermore, the timing of sample collection in suspected germline TP53 carriers plays a significant role in result interpretation, as somatic TP53 is one of the most common CH lesions [45, 56,57,58], In the paired blood-tumor analysis from MSK-IMPACT the samples obtained from older patients, or following chemotherapy administration were found to have higher rate of somatic TP53 mutations most likely representing chemotherapy-included CH rather than germline TP53 variants [53, 55].
In recently published analysis of 140,597 participants without hematologic neoplasms from BioBank Japan, TP53-CH was associated with poor overall survival (HR 1.42) [59]. Not unexpectedly, individuals with TP53-CH were found to have increased mortality from myeloid and lymphoid hematologic malignancies compared to non-carriers. This adverse prognostic impact was evident even among those without prior malignancies, indicating that the risks is not solely attributable to secondary myeloid neoplasms [
Comments (0)