
Remember me
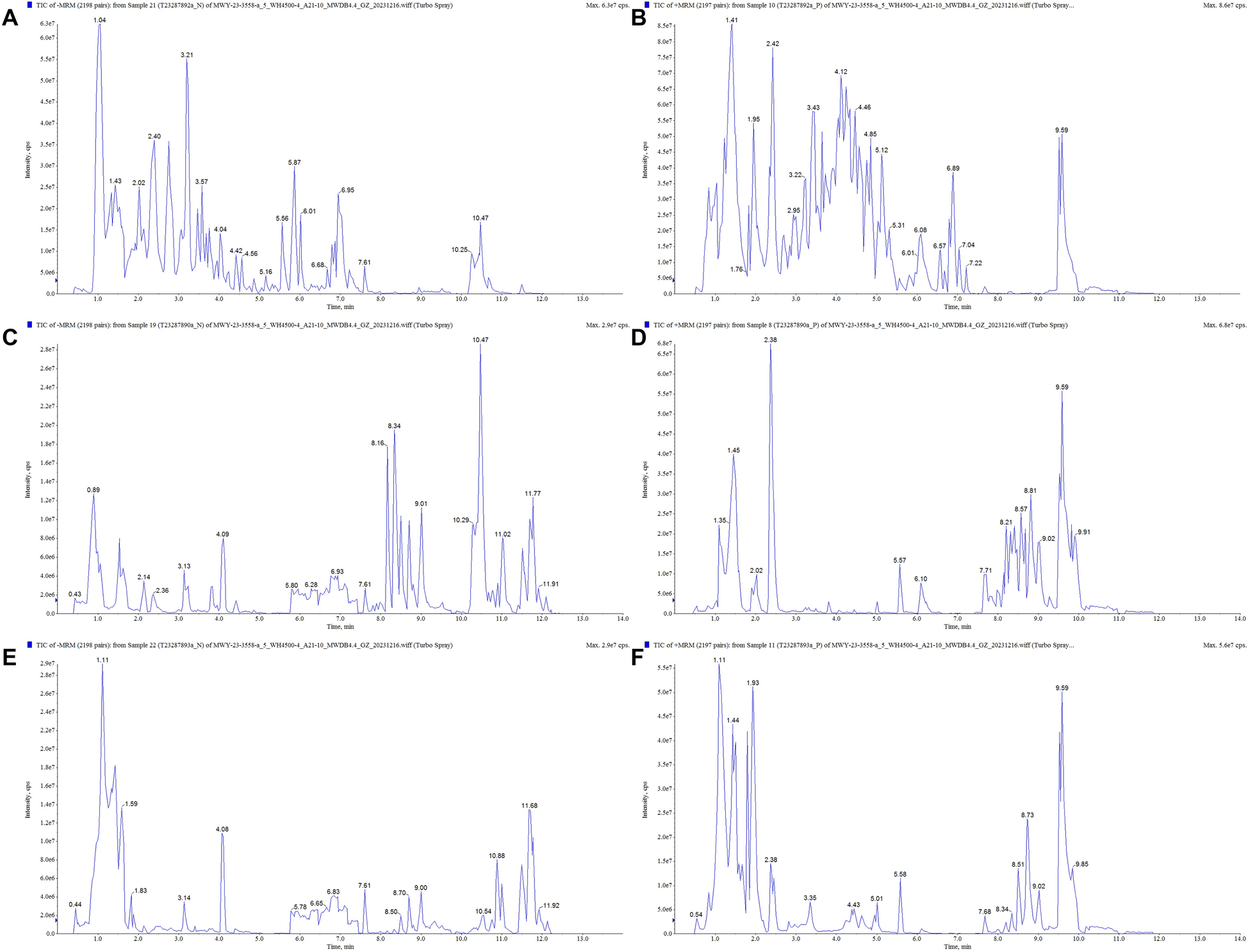
AR is the only traditional Chinese medicine contained in AROL. The UHPLC-Q-TOF/MS TICs of AROL in the positive and negative ion modes are shown in Figure S1. Totally 69 representative compounds were finally identified, and their detailed information was summarized in Table S2. Of these, 23 compounds were subsequently validated through comparison with the corresponding standards. The component analysis results clearly demonstrated that AROL was mainly composed of AR-derived flavonoids, saponins, amino acids, and polysaccharides.
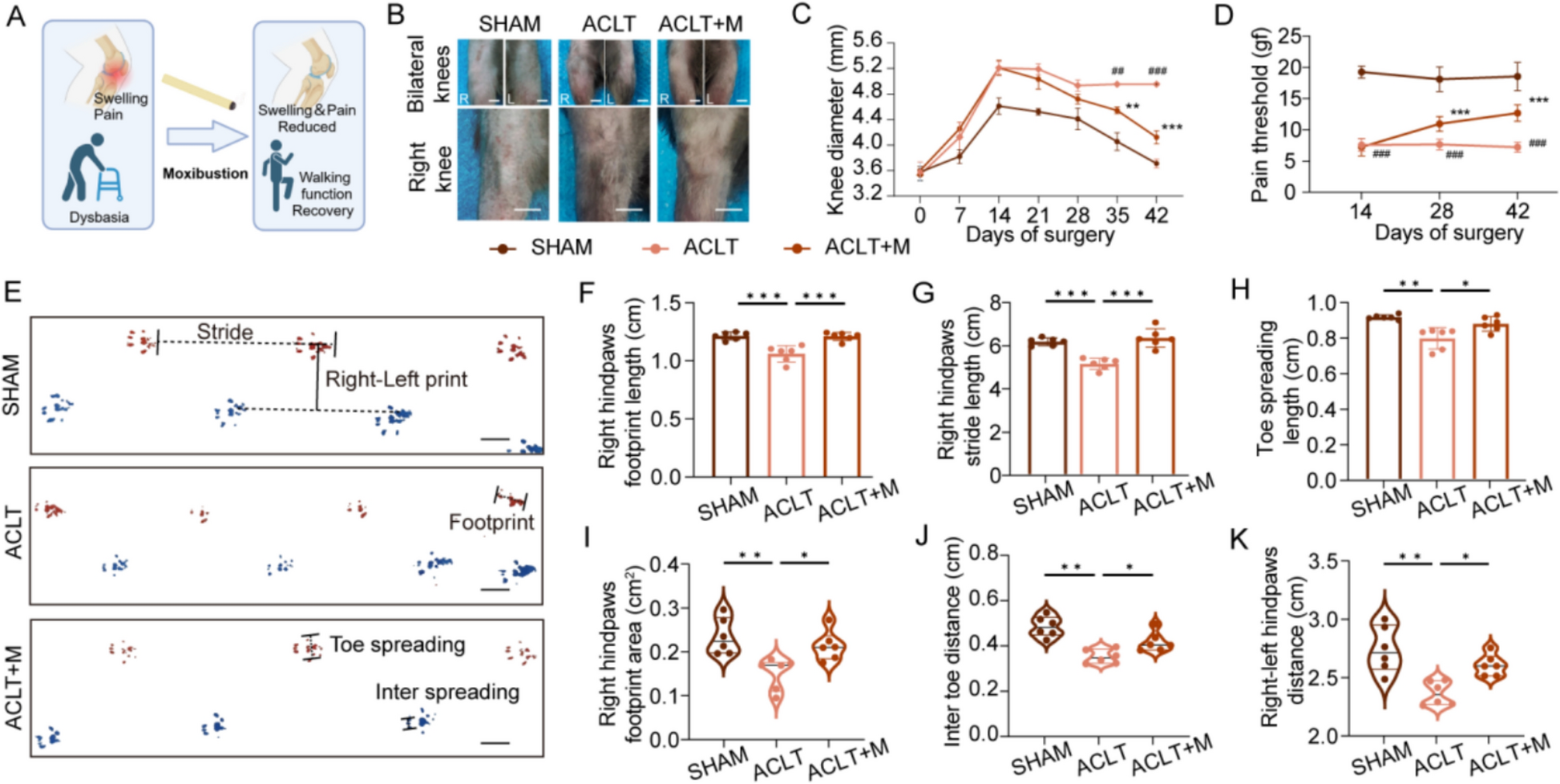
Clinical pharmacological assessmentA total of twenty normal weight (BMI: 21.7 ± 1.7 kg·m−2) healthy adults between 24–36 years old were recruited to the protocol. All twenty subjects completed this study with high compliance and no significant changes in their dietary patterns were observed throughout the intervention period. Their blood pressures including SBP and DBP were also measured, and the SBP/DBP in Group d0 was 116.7 ± 8.7/72.2 ± 7.5 mmHg and 118.1 ± 8.5/75.10 ± 8.0 mmHg in Group d15. No notable differences were observed in physiological functions or liver function between Group d0 and Group d15 (Table S3), which confirmed that no serious adverse events were observed in subjects after AROL administration. However, Scr in renal function showed a significant decrease in Group d15.
Two types of IIs, cells, and cytokines, were then used to assess the modulation of human immunity after the administration of AROL. As for blood immune cells (Figure S2), there was an increase in the levels of CD4+ and CD8+ T cells, while a decline was observed in CD19+ (B cells) and CD3-CD16+CD56+ (NK cells) after AROL administration, however, these changes in immune cells showed no statistical significance. Unlike this, most of the immune cytokines were greatly affected by AROL administration. As shown in Fig. 2, five immune cytokines (IFN-γ, TNF-α, IL-4, IL-6, and IL-12P70) were significantly up-regulated in Group d15, while IL-2 was significantly down-regulated. IL-10 and IL-17 also showed a decreased trend but no significant change.
Fig. 2
The modulation of human immunity after the administration of AROL. A Measurement of serum creatinine (Scr) in two groups. The impact of AROL on immune cytokines B TNF-α, C IFN-γ, D IL-2, E IL-4, F IL-6, G IL-12P70, H IL-10, and I IL-17. (*p < 0.05; **p < 0.01; ***p < 0.001, n.s means non-significant.)
Metabolomics and lipidomics analysisData quality assessmentAs shown in Figure S3, the TICs of all the pooled QC samples displayed excellent overlap throughout each analytical run. This result indicated the instrumental system maintained high consistency during sample analysis, ensuring the collected metabolomics and lipidomics datasets from serum, urine, and feces were of superior quality. Meanwhile, throughout each analysis, the RSD% values for the m/z and RT of five characteristic features remained below 0.001% and 0.7%, respectively, and the RSD% values for intensity were consistently under 30% (Table S4), further indicating that the instrumental stability and repeatability were acceptable [18]. It should be noted that the cluster of QC samples in our original data was not always good enough, just as shown in Figure S4, suggesting that there may be some systematic error in these ten runs. SERRF is a normalization method based on QC samples, which could eliminate unnecessary system changes in large sample sets [14]. After being SERRF-normalized, the QC clustering was significantly improved (Fig. 3), clearly demonstrating the suitability and potential of SERRF normalization, so the SERRF-normalized data was adopted of all ten runs for further analysis in our study. Collectively, the raw data collected by our current RPLC/HILIC-Q-TOF/MS methods was demonstrated to be reliable and could reflect the real metabolic condition of the corresponding biological samples.
Fig. 3
The relatively subtle impact of AROL administration on the metabolic profiles of healthy adults. PCA score plots (after SERRF) of Group d0 and Group d15: A Serum LA pos; B Serum LA neg; C Serum MA pos; D Serum MA neg; E Urine pos; F Urine neg; G Feces LA pos; H Feces LA neg; I Feces MA pos; J Feces MA neg. LA Lipidomics analysis, MA Metabolomics analysis, pos positive mode, neg: negative mode
Metabolic profiling analysisAs an unsupervised analytical approach, PCA offers an impartial overview of the metabolic profiles for all samples. Visual inspection of PCA score plots in both LA and MA (Fig. 3) showed no obvious difference between Group d0 and Group d15, indicating AROL administration has a relatively subtle impact on the metabolic profiles of healthy adults. This could partially establish a solid foundation for considering AROL as a nutritional supplement. PLS-DA was subsequently employed to further investigate the variations in metabolic profiles between the groups. The PLS-DA score plots (Figure S5) show that the subjects could be divided into two distinct clusters corresponding to Group d0 and Group d15, demonstrating that successive AROL administration could produce some metabolic changes. However, not all the PLS-DA models could meet the given acceptance criteria in “Data analysis” section, and especially Q2Y values in the PLS-DA models from urine and feces were all less than 0.25, indicating there may be large and uncontrollable individual differences in the clinical samples. Accordingly, VIP values from PLS-DA were not used for the following feature screening. Instead, the two-tailed paired t-test and FC analysis were adopted to filter out the AR-induced SMFs (Figure S6). Table S5 summarizes the changes in the number of features during the entire data processing of LA and MA. In serum and feces samples, the number of features was high, and the identified PBs including different lipid and polar compounds by performing LA and MA respectively, which confirmed the necessity of our analysis using a combination of RPLC-Q-TOF/MS and HILIC-Q-TOF/MS techniques.
Serum lipidomics and metabolomics analysis1166 and 1752 features were obtained from serum LA and MA respectively following MS-DIAL data preprocessing and MS-FLO data cleaning, from which a total of 209 and 82 SMFs were screened out according to the criteria of p < 0.05, and FC > 1.2 (or < 0.83). 51 and 38 differential metabolites were finally identified as AR-induced serum PBs, and their detailed information including tR, m/z, ion type, formula, name, PubChem CID, HMDB ID as well as p and FC values from group comparison, were shown in Table S6.
Metabolic pathway enrichment analysis showed that only 11 (out of 51) LA-derived serum PBs could be associated with valid KEGG IDs. As a result, six pathways were enriched and each of these pathways was linked to lipids (Fig. 4A1). The major enriched pathways (p < 0.05) were linoleic acid metabolism, alpha-linolenic acid metabolism, glycerolipid metabolism, and sphingolipid metabolism. These pathways were enriched by three classes of metabolites, phosphatidylcholines (PC), TG, and sphinganine, which were all significantly up-regulated in the serum of Group d15. Up to 17 (out of 38) MA-derived serum PBs were found to be enriched for valid KEGG IDs, and consequently as many as 17 metabolic pathways were enriched (Fig. 4A2). Of them, the major pathways (p < 0.05) were arginine and proline metabolism and linoleic acid metabolism, with increased aminobutyric acid, L-proline, ornithine, and phosphatidylcholine in Group d15.
Fig. 4
Identification and pathway analysis of serum, urine, and feces metabolites. A The pathway enrichment analysis of AROL-induced PBs. B Chemical similarity enrichment analysis of the PBs. Cluster colors give the proportion of increased or decreased compounds (red = increased, blue = decreased, purple = both increases and decreases) in each cluster. Dots size indicates the number of PBs in each cluster. The top of the Y-axis shows the most obvious clusters. (1) serum, LA; (2) serum, MA; (3) urine; (4) feces, LA; (5) feces, MA. LA Lipidomics analysis, MA Metabolomics analysis
Since only 11 (out of 51) LA-derived serum PBs could be enriched with effective KEGG IDs, the metabolite enrichment analysis with ChemRICH was further performed. Data matrices of serum PBs were then subjected to ChemRICH analysis for the determination of chemical similarity enrichment of the PBs. Five clusters including triglycerides (TG), glycerophospholipids, diglycerides (DG), and ceramides (Cer) were enriched in the ChemRICH analysis of 51 serum PBs from LA (Fig. 4B1), and these clusters were significantly up-regulated in Group d15. As for 38 serum PBs from MA, only two clusters were enriched: Saturated Fatty Acids (SFAs) with an up-regulated tendency, and phosphatidic acids with a significant down-regulation in Group d15 (Fig. 4B2).
Urine metabolomics analysisA total of 1752 features were detected in the MA, with 113 of them were screened out as SMFs. Finally, 41 differential metabolites were identified as AR-induced urine PBs, and their detailed information was summarized in Table S7. 26 of 41 identified urine PBs were found to be related to 9 metabolic pathways by pathway enrichment analysis (Fig. 4A3). Of them, the major pathways (p < 0.05) were aminoacyl-tRNA biosynthesis and nicotinate and nicotinamide metabolism, with an increase in l-arginine and a decrease in nicotinamide, N1-methyl-2-pyridone-5-carboxamide and l-methionine in Group d15. Moreover, three of the compounds were enriched to the cluster of pyridines (Fig. 4B3) through ChemRICH analysis, with a significant decrease in nicotinamide and N1-Methyl-2-pyridone-5-carboxamide and a significant increase in nitrendipine in Group d15.
Feces lipidomics and metabolomics analysisAfter preliminary data processing by MS-DAIL and MS-FLO, 1968 and 2059 features were present in feces LA and MA, respectively, from which 198 and 82 SMFs were screened. Of them, 64 and 40 metabolites were finally identified as AR-induced feces PBs, and their detailed information was shown in Table S8. Significantly, only 27 out of the 64 feces PBs were enriched with valid KEGG IDs in LA, resulting in the enrichment of a total of 12 metabolic pathways through pathway enrichment analysis (Fig. 4A4). Five major enriched pathways (p < 0.05) were sphingolipid metabolism, glycerophospholipid metabolism, phenylalanine, tyrosine, and tryptophan biosynthesis, linoleic acid metabolism, and phenylalanine metabolism. These pathways were enriched by five classes of metabolites including SM, galactosylceramide, glucosylceramide, PC, and phenylalanine which were all significantly down-regulated in the feces of Group d15. Up to 19 (out of 40) MA-derived feces PBs could be enriched for valid KEGG IDs, as well as 5 metabolic pathways were enriched (Fig. 4A5). The major enriched pathway (p < 0.05) was vitamin B6 metabolism, with increased 2-Oxo-3-hydroxy-4-phosphobutanoic acid in Group d15.
According to ChemRICH analysis, 64 feces PBs from LA could be roughly divided into eight clusters, including PC, sphingomyelins (SM), saturated_lysophospholipids, sitosterols, TG, galactosylceramides, benzene derivatives, and oleic acids (Fig. 4B4). These clusters were all significantly down-regulated in Group d15. As for 40 feces PBs from MA, two clusters were enriched, DG and UFA both with an up-regulated tendency in Group d15 (Fig. 4B5).
Comprehensive analysis of serum, urine, and fecesUp to 234 AR-induced PBs were screened and identified from LA and MA, of which 89, 41, and 104 were in serum, urine, and feces, respectively. The identification of such a large number of PBs partly indicated that AR could regulate human metabolic profiles. Further analysis (Figure S7) showed that the median FC-values of these PBs in three samples after AR administration were all less than 2, and the median p-values were no more than 0.025, clearly demonstrating the mild regulatory effect of AR on human metabolome and lipidome. Chemical structure enrichment analysis of PBs resulted in 18 clusters, including 7, 1, and 10 from serum, urine, and feces. A total of 49 metabolic pathways were enriched by pathway enrichment analysis using Metaboanalyst, of which 23, 9, and 17 were from serum, urine, and feces. Venn analysis among serum, urine, and feces was performed for AR-induced PBs, their related pathways, and enriched compound clusters to find the common changes in all three biological samples. As shown in Figure S8A, no shared identified PBs were observed in the three samples, reflecting the advantage of the comprehensive metabolomic analysis performed in this study. 2-Benzylsuccinic acid was present in both urine and feces. Four lipid PBs, namely SM, Pantothenol, TG, and DG, were common in serum and feces. Besides, up to 99 PBs were only detected in feces, 85 PBs were merely present in serum and 51 PBs were unique in urine, fully demonstrating the necessity of simultaneous analysis of various biological samples.
Figure S8B illustrates that there are four pathways (purine metabolism, cysteine and methionine metabolism, arginine and proline metabolism, and arginine biosynthesis) were common in both serum and urine, and six pathways (glycerolipid metabolism, alpha-Linolenic acid metabolism, arachidonic acid metabolism, sphingolipid metabolism, glycerophospholipid metabolism, and linoleic acid metabolism) were common in serum and feces. Only aminoacyl-tRNA biosynthesis was simultaneously enriched in all three samples, corresponding to three amino acid metabolites including L-phenylalanine, L-proline, and L-arginine. Therefore, phenylalanine metabolism and arginine metabolism are considered to be the predominant metabolic pathways. Since many lipids within the same class often have a shared KEGG ID, the pathway enrichment analysis feature in MetaboAnalyst was not particularly effective for annotating lipid metabolites. In contrast, ChemRICH was able to partially enhance the understanding of the structural grouping and relationships among the lipid PBs. As shown in Figure S8C, three clusters (Cer, TG, and DG) were common in serum and feces. It is evident that there are more lipid metabolites and lipid metabolism pathways shared in serum and feces, and thus lipid metabolism pathways also serve as priority pathways regulated by AR. Therefore, it could be speculated that amino acid metabolism and lipid metabolism are the main metabolic pathways by which AR regulates human immunity.
Gut microbiome analysisCharacterization of gut microbiotaIn this study, feces samples were used to evaluate the improvement of AROL on the human gut microbiota. The 16S rRNA sequencing yielded a total of 4,142,541 optimized sequences. After being filtered by a 97% identity threshold, a total of 1077 OTUs was obtained for further gut microbiome analysis. Both the species accumulation curves and the rarefaction curves demonstrated that the sequencing depth, data quality, and the sample size were sufficient (Figure S9).
Species diversity analysisAs shown in Fig. 5A, Venn analysis revealed that Group d0 had 140 unique OTUs and Group d15 had 219 distinct OTUs, while up to 718 OTUs (accounting for 2/3 of the total OTUs) were common between the two groups, indicating that Group d15 possessed a slightly higher richness of gut microbiota than Group d0. Similarly, the alpha diversity analysis revealed no significant changes between Group d0 and Group d15, with the corresponding p-values of 0.810, 0.879, 0.395, and 0.348 for Chao, ACE, Shannon, and Simpson indices (Figure S10). The results revealed that the administration of AROL did not produce notable alterations in the abundance or diversity of the microbial community, indicating that AROL has a limited impact on the gut microbiome of healthy adults. To illustrate the effect of AROL administration on species composition, bar plots were employed at the phylum and genus taxonomic levels. As displayed in Fig. 5B, Firmicutes, Actinobacteria, Proteobacteria, and Bacteroidota were the dominant bacterial phylum in both groups and accounted for more than 95% of the bacteria in the microbial communities. In Group d15, Firmicutes showed a downward trend while Actinobacteriota displayed a clear upward trend. At the genus level, the fecal microbiome was dominated by Collinsella, Bifidobacterium, Blautia and Eubacterium_hallii_group, and Subdoligranulum (Fig. 5C). We further compared the abundance differences of the top three genera with high abundance (Collinsella, Bifidobacterium, and Blautia) between two groups. In Group d15, the abundance of Collinsella and Bifidobacterium exhibited an upward trend, whereas the levels of Blautia displayed a downward trend (Fig. 5D).
Fig. 5
AROL regulated gut microbiota of healthy humans. A Shared and unique microbiota between Group d0 and Group d15. The structural composition of the gut microbiota in each group at the B phylum level and C genus-level (relative abundances are reported on the vertical axis). D Changes in the abundance of Collinsella, Bifidobacterium, and Blautia after AROL administration. E NMDS analysis on OTU level. F PLS-DA analysis on OTU level. G The difference in abundance at the phylum level and the genus level in Group d0 and Group d15. (*p < 0.05; **p < 0.01; ***p < 0.001)
Bacterial differences analysisAs shown in Fig. 5E, the clustering of the two groups in the NMDS plot was nearly overlapped (p = 0.954), indicating the similar community composition between Group d0 and Group d15. However, the PLS-DA score plot revealed a clearer distinction between the two groups, which indicated that AROL exhibited significant modulatory effects on gut microbial community structure (Fig. 5F). Figure 5G showed that there are considerable variations in the abundance of phylum and genus between Group d0 and Group d15. Especially at the phylum level, a marked reduction in the abundance of Bacteroidota was observed in Group d15. At the genus level, administration of AROL resulted in significant changes in six genera. Specifically, Anaerofustis and Enorma exhibited substantial increases, whereas Prevotella, Granulicatella, Corynebacterium, and Parasutterella were notably reduced in Group d15.
Spearman’s correlation analysisSpearman’s correlation analysis was applied to explore the relationship among the changes in metabolic profiles, gut microbes, and significant IIs.
Seven significant IIs were attempted to be correlated with six differential gut genera and twenty major abundant genera, respectively. As shown in Fig. 6A, of all the six different genera, only Granulicatella was negatively correlated with TNF-α and IL-4, and there was no more correlation between the remaining genera and significant IIs. Further correlation analysis between the differential genus Granulicatella and PBs, revealing a positive correlation with GlcCer (p < 0.05), SM (p < 0.05), and Lyso Phosphatidylethanolamine (LysoPE) (p < 0.05), a negative correlation with L-arginine (p < 0.01) and DG (p < 0.05). As shown in Fig. 6B, nine of twenty major genera were significantly correlated with at least one of the immune cytokines. Notably, Bifidobacterium was correlated with three indicators, including a positive correlation with IL-4 (r = 0.555, p = 0.002), IL-12P70 (r = 0.524, p = 0.004), and TNF-α (r = 0.381, p = 0.046). Additionally, Scr and five genera were significantly correlated, including a negative correlation with Dialister (r = − 0.466, p = 0.012), Collinsella (r = − 0.548, p = 0.002), Blautia (r = − 0.439, p = 0.019), and Faecalibacterium (r = −0.521, p = 0.004) and a positive correlation with Romboutsia (r = 0.393, p = 0.039). These findings suggested that the immunomodulation of AR may be closely associated with major genera rather than differential microbiota. In order to explore the possible microbiota-mediated metabolic mechanisms for the immunomodulation of AR, the top 20 major genera and seven significant IIs were further used to be correlated with AR-induced PBs from different biological samples.
Fig. 6
Spearman’s correlation analysis between genera and significant immune indicators (IIs). A Between 6 differential genera and seven significant IIs. B Between the 20 major genera of abundance and seven significant IIs. (*p < 0.05; **p < 0.01; ***p < 0.001)
AR-induced serum PBs correlation analysis demonstrated that (Fig. 7A) there were strong correlations between six major microbiota genera including Bifidobacterium, Anaerostipes, Streptococcus, Clostridium, Erysipelotrichaceae, and Fusicatenibacter, and four types of serum PBs including glycerides (eg., TG), sphingolipids (eg., Cer), FA, and carnitines. Generally, 81% of serum PBs and gut microbiota are directly correlated (Fig. 7D). Typically, TG showed a strong positive correlation with Bifidobacterium (p < 0.01) and Streptococcus (p < 0.05), while l-Proline was positively correlated with Ruminococcus (r = − 0.476, p = 0.010). In addition, Bifidobacterium was also positively correlated with Insoine (r = 0.489, p = 0.006) and polyunsaturated fatty acids (PUFAs) (p < 0.05). There were also significant correlations between serum PBs and significant IIs, and especially, Scr showed a strong positive correlation with Cer (p < 0.05).
Fig. 7
Spearman’s correlation analysis of 7 immune indicators, 20 major genera, and AR-induced PBs. Hierarchical clustering analysis between AR-induced PBs from A serum, B urine, and C feces and gut microbiota, as well as immune indices; red represents a positive correlation, while blue represents a negative correlation, the darker color indicates a stronger correlation (*p < 0.05; **p < 0.01; ***p < 0.001). The pie chart of AR-induced PBs from D serum, E urine, and F feces related to gut microbiota
AR-induced urine PBs correlation analysis (Fig. 7B) demonstrated that there was a strong correlation between two genera including Erysipelotrichaceae_UCG-003 and Subdoligranulum, and two PBs including Niacinamide and Methylglutaric acid. Erysipelotrichaceae_UCG-003 was negatively correlated with Niacinamide (r = − 0.538, p = 0.003). Although only two urine PBs had a strong correlation with genera, up to 84% of urine PBs correlated with gut microbiota (Fig. 7E). In addition, there were also significant correlations between urine PBs and significant IIs, for example, IL-2 was positively correlated with ethosuximide (r = 0.535, p = 0.002) and acoric acid (r = 0.583, p < 0.001).
AR-induced feces PBs correlation analysis (Fig. 7C) demonstrated that there were six genera including Bifidobacterium, Erysipelotrichaceae, Faecalibacterium, Ruminococcus, Streptococcus, and Subdoligranulum correlated strongly with five types of feces PBs including SCFAs (eg., Isobutyric acid), Phenylalanine (eg., L-phenylalanine, 2-Phenylpropionate), glycerides (eg., DG, TG), glycerophospholipids (eg., LysoPE, PC), and sphingolipids (eg., SM, Cer). Overall, gut microbiota and 65% feces PBs were correlated (Fig. 7F). Ruminococcus and SM (r = 0.55, p = 0.005) showed a positive correlation while Subdoligranulum and Isobutyric acid (r = − 0.439, p = 0.033) showed a negative correlation. Notably, Bifidobacterium showed negative correlations with GlcCer (p < 0.05) and L-phenylalanine (r = − 0.537, p = 0.035). Meanwhile, Scr showed a strong negative correlation with Isobutyric acid (r = − 0.527, p = 0.008) and SM (p < 0.01), while TNF-α (p < 0.05) and IFN-γ (p < 0.05) showed strong positive correlations with Cer. In addition, PC showed strong negative correlations with IL-2 (p < 0.05), IL-4 (p < 0.05), and TNF-α (p < 0.05).
Comments (0)