Cell culture
FaDu (hypopharyngeal carcinoma cell line) was obtained from American Type Culture Collection (ATCC, Manassas, VA), while HK-1 (nasopharyngeal carcinoma cell line) was donated by Professor George Tsao, University of Hong Kong [33]. FaDu cells were cultured in MEM medium (Gibco) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin (100U/mL). HK-1 cells were cultured in RPMI-1640 medium (Biowest) with the addition of 1% sodium pyruvate, 1% MEM Non-Essential Amino Acids solution (NEAA), 1% L-glutamate, 10% FBS, and penicillin/streptomycin (100U/mL). All cell cultures were maintained at 37 °C under a humidified atmosphere with 5% CO2. All cell lines used in this experiment were authenticated by ATCC cell line authentication service.
Generation and characterization of in vitro RR-HNC models
RR FaDu and HK-1 cell lines were generated by exposing their respective parental cells to a course of fractionated 2 Gy irradiation over 45 daily treatments, five times a week, to a total dose of 90 Gy. Cells were seeded in a 175 cm2 flask prior to irradiation. Irradiation was delivered using a Gamma Cell®GC40 exactor source 137Cs-137 (Nordion, Canada) at a dose rate of 0.9 Gy/min. Routine media change of the irradiated cells was performed during the course of irradiation and passaged at 80% confluency. The induction of radiation resistance in the RR sublines was confirmed using clonogenic assays and characterization of its biological properties were performed using proliferation and migration assays (Fig. S1).
Proliferation assay
FaDu and HK-1 cells were seeded at low density (500 cells/well) in 384-well plates and allowed to attach for 3 h. Cell viability was determined by CellTiter-Glo (CTG) Luminescent Cell Viability Assay (Promega) according to manufacturer’s instructions every 24 h for a total of 96 h. Values obtained in Relative Light Units (RLU) were normalized to media-only control wells and fitted to a growth curve using Prism 9 software (GraphPad).
Migration assay
Serum-starved FaDu and HK-1 cells were plated in 6-well plates (1 × 106 cells/well) and allowed to recover overnight. A sterile 200 μL pipette tip was used to make a vertical scratch in the cell monolayer, followed by two wash steps to remove debris from the edge of the scratch. Acquisition of images were performed at indicated timepoints with a 10 × phase objective using an Olympus IX71 microscope (Olympus Canada, Richmond Hill, Ontario). Wound healing capacity was determined using ImageJ wound healing tool and plotted with Prism 9 software (GraphPad).
Determination of half-maximal inhibitory concentration (IC50) of drugs
Cells were seeded in 384-well plates (2500 cells/well) 24 h prior to drug treatment. Drugs were prepared in serial dilutions that ranged from 0.0001 μM to 100 μM and cells were treated in technical quadruplicates for 48 h. Cell viability was then measured using CTG assay (Promega) according to manufacturer’s instructions. IC50 values of individual drugs were determined using Prism 9 software (GraphPad) by fitting normalized cell viability values into sigmoidal dose–response curves.
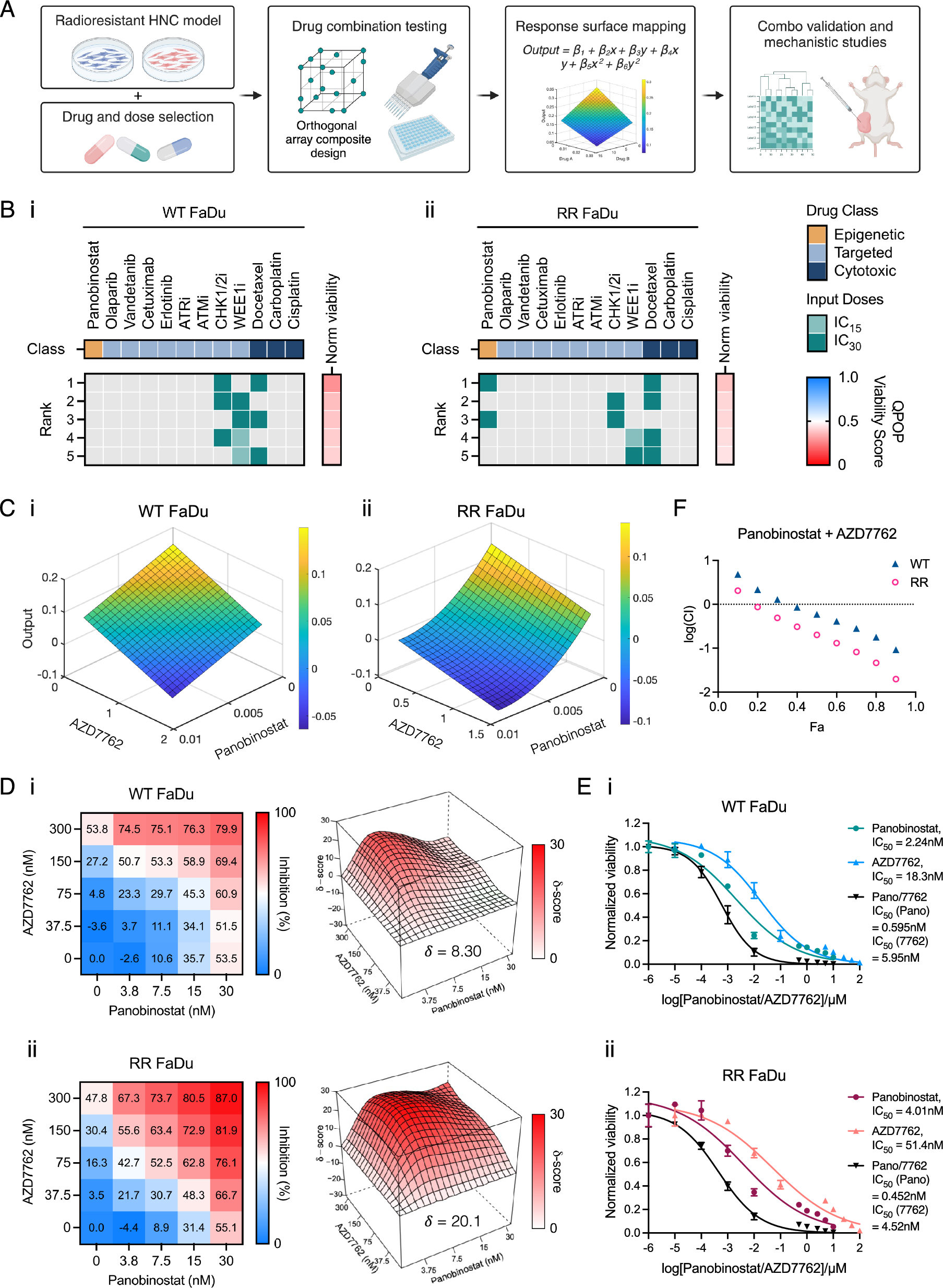
QPOP combinatorial drug treatment and analysis
A 12 drug-three dosages (IC0, IC15, IC30) QPOP was performed in this study. The drug panel included cytotoxic chemotherapy agents (docetaxel, cisplatin, carboplatin), epidermal growth factor receptor (EGFR) (cetuximab, erlotinib) and DNA damage response (DDR) targeted therapies (PARP1/2, ATM, ATR, CHK1, WEE1 inhibitors), an HDAC inhibitor (panobinostat) and a multi-tyrosine kinase inhibitor (vandetanib). Both WT and RR FaDu lines were screened against log dose concentrations of the panel of 12 drugs for 48 h to establish their respective dose–response curves and half-maximal inhibitory concentrations (IC50). IC15 and IC30 concentrations were intrapolated from the dose–response curves. An orthogonal array composite design (OACD) was used as previously described to determine the 155 experimental combinations necessary for sufficient factor screening and model fitting, and tested in technical duplicates [34]. Drugs were dispensed with the aid of a digital dispenser (D300e, Tecan) and cell viability was measured 48 h post drug treatment using CTG assay (Promega). The normalized cell viability scores were used as phenotypic inputs for QPOP second-order regression analysis as described by Rashid et al. [24]. QPOP then generates corresponding viability output values for all possible drug permutations. All two-drug combinations were subsequently ranked according to the output and parabolic response surface maps indicating interaction between any two drugs in combination were derived.
Validation of drug combinations
To validate the top-ranked drug combinations determined by QPOP, we employed the use of mathematical models to quantify the combination effect of two drugs and provide better interpretation and assess reproducibility of QPOP-derived experimental data. In the median-effect (Chou-Talalay) method, single-drug and combination dose–response assays were performed to determine the IC50 values of each drug as monotherapy and in combination. The ratio of drug dosages between the two drugs was determined by the dosages used in QPOP and kept constant across the range of concentrations treated. Cell viability was then measured using CTG assay (Promega) 48 h post drug treatment and the combination index (CI) values were calculated according to the Chou-Talalay method [35]. Additionally, the Bliss independence model was used to compute the degree of drug synergy by categorizing combinations as synergistic, antagonistic or additive. Serial dilutions of either drug were prepared in deep well plates and added to cells in a 5 × 5 matrix of varying drug doses, with technical quadruplicates. Cell viability was measured after 48 h using CTG Assay (Promega). Bliss synergy scores were computed by inputting normalized cell viability values into SynergyFinder 2.0 application [36]. Based on deviation of observed and expected responses in the Bliss independence model, the drug combination interaction is classified as synergistic (> 10) or antagonistic (< − 10).
Apoptosis assay
Following 48 h of drug treatment, cells were harvested and apoptosis levels were determined using the BD Pharmingen™ FITC Annexin V Apoptosis Detection Kit as per manufacturer’s instructions (BD Biosciences). Briefly, cells were stained with FITC Annexin V antibody and Propidium Iodide (PI) and percentage of cells undergoing apoptosis was measured by flow cytometry (BD LSRII, BD Biosciences). At least 10,000 events were analyzed for each sample. Quantification of percentage of apoptotic cells was performed using FlowJo software.
Comet assay
Following 48 h of drug treatment, cells were harvested and mixed with 1% LMAgarose in PBS at 37 °C. Cell suspensions, adjusted to 10,000 cells/mL, were layered onto precoated microscope slides and allowed to solidify at 4 °C for 10 min. The slides were then immersed in lysis buffer (100 mM disodium EDTA, 2.5 M NaCl, 10 mM Tris–HCl, pH 10) containing 1% Triton X-100 overnight at 4 °C. The slides were subsequently placed in alkaline electrophoresis buffer (300 mM NaOH, 1 mM disodium EDTA, pH > 13) for 45 min to allow unwinding of the DNA. Single cell gel electrophoresis was conducted in the same buffer on a horizontal electrophoresis platform for 20 min at 25 V, 400 mA. Lastly, the slides were rinsed in neutralizing buffer (0.4 M Tris–HCl, pH 7.5), followed by staining with PI. Images were visualized by a confocal laser scanning microscope (LSM 880 Airy Scan, Carl Zeiss), with at least 5 images per sample and tail: head ratio was determined by the OpenComet software.
Clonogenic assay
FaDu and HK-1 cells were seeded in a 60 mm dish and treated with 10 nM or 20 nM panobinostat for 24 h. Cells were subsequently irradiated with a series of 1, 2, and 4 Gy doses, and reseeded in 6-well plates. After 10–14 days, cells were stained with 0.05% crystal violet for 1 h. Colonies with more than 50 cells were counted with a microscope. Plating Efficiency (PE) was calculated by the ratio of the number of colonies counted and the number of cells plated. The surviving fraction (SF) was calculated by the ratio between the PE of irradiated cells and the PE of the non-irradiated cells. Prism 9 software (GraphPad) was used to generate the survival curves using the non-linear regression LQ model (Y = EXP [-(B1*X + B2*X^2)]).
siRNA-mediated knockdown
FaDu and HK-1 cells were transfected with si-Negative Control (si-NC) or respective siRNAs (TriFECTa® DsiRNA Kit, Integrated DNA Technologies, IDT, Singapore) using Lipofectamine RNAiMAX (Thermo Fisher) following the manufacturer’s instructions. For combinatorial drug dosing studies, AZD7762 (MedChemExpress, HY-10992) was added to cells 24 h post-transfection. Cell viability was measured 48 h post drug treatment using CTG assay (Promega), or cells were pelleted at the specified timepoints and used for protein extraction as described below. Knock-down efficiency was evaluated 72 h after transfection using western blot.
Western blot
Cells were pelleted and washed with cold phosphate buffered saline (PBS) twice before being lysed in RIPA lysis buffer (ThermoFisher Scientific) containing both phosphatase (PhosSTOP; Roche) and protease inhibitors (cOmplete™ Protease Inhibitor Cocktail; Roche). The lysates were collected, centrifuged at 12,000 × g for 10 min, and respective protein concentrations were determined with a bicinchoninic acid protein (BCA) assay kit (Pierce, Iselin, NJ, USA). Equal amounts of protein lysates were resolved by SDS-PAGE, transferred onto PVDF membranes and serially stained with primary and secondary HRP-conjugated antibodies according to standard procedures. Protein bands were then detected via chemiluminescence using the ChemiDoc Imaging system (Bio-Rad). Primary antibodies used include PARP (Cell Signaling Technology, #9542), Caspase-3 (Cell Signaling Technology, #9662), cleaved Caspase-3 (Cell Signaling Technology, #9664), acetylated-lysine (Cell Signaling Technology, #9814), RAD51 (Abcam, ab133534), γ-H2A.X Ser139 (Cell Signaling Technology, #9718), c-MYC (Abcam, ab32072), p21 Waf1/Cip1 (Cell Signaling Technology, #2947), HDAC1 (Cell Signaling Technology, #5356), HDAC2 (Cell Signaling Technology, #5113), HDAC4 (Cell Signaling Technology, #7628), HDAC6 (Cell Signaling Technology, #7558), acetylated-α-tubulin Lys40 (Cell Signaling Technology, #3971), acetyl histone H3 Lys27 (Cell Signaling Technology, #8173), GAPDH (Cell Signaling Technology, #2118), and SP1 (Millipore, #07-645).
Co-immunoprecipitation (co-IP) assay
Pelleted cells were washed with cold PBS twice, resuspended in 500μL lysis buffer (50 mM Tris, pH 7.6; 150 mM NaCl; 0.5% Triton-X100; 0.1% Nonidet P-40) containing protease inhibitors (Roche) and incubated on a rotator for 30 min at 4 °C. Cell lysates were centrifuged at 12,000 × g for 15 min, and protein concentrations were determined using BCA assay and normalized across samples. 800 µg of lysates were incubated with 5 µg anti-SP1 (Millipore, #07–645), anti-acetylated-lysine (Cell Signaling Technology, #9814) or normal rabbit IgG (Cell Signaling Technology, #2729) overnight at 4 °C. 25μL of Dynabeads™ Protein G (Invitrogen, 10003D) were then added to lysates and left for 2 h at 4 °C under rotation. After magnetic immunoprecipitation and washes, immunoprecipitates were resolved by SDS-PAGE, transferred, and blotted as described above.
Electrophoretic mobility shift assay (EMSA)
Nuclear protein fractions were isolated using the NE-PER™ Nuclear and Cytoplasmic Extraction Kit (ThermoFisher Scientific) following manufacturer’s protocol. A non-radioactive electrophoretic-mobility shift assay (EMSA) kit (Signosis, GS-0040) was used to assess the transcription binding activity of SP1. Briefly, nuclear extract (10 μg) was incubated with 1 µl poly D (I-C), 2.0μL 5X Binding Buffer and 1.0μL of SP1 biotin-labelled probe or cold probe at 22 °C for 30 min in a thermocycler. DNA–protein complexes were electrophoresed in a 6.5% non-denaturing polyacrylamide gel and transferred to a positively charged nylon membrane. The protein-bound probe and free probe were immobilized with UV cross-linker and the biotin end-labelled probe was detected with streptavidin-HRP via chemiluminescence.
Chromatin immunoprecipitation (ChIP) and quantitative PCR
Cells were washed with cold PBS twice, crosslinked with 1% (v/v) formaldehyde (Pierce) for 15 min and quenched with 0.125 M glycine. The fixed cells were lysed and sonicated for 15 cycles (15 s ON/30 s OFF) at 30% amplitude using EpiShear Probe Sonicator (Active Motif). 25 μg of sheared chromatin was incubated with 5 μg of antibodies overnight at 4 °C under rotation. 25μL of pre-equilibrated Dynabeads™ Protein G (Invitrogen, 10003D) was added to the ChIP reactions the next day and left to conjugate for 2 h at 4 °C. Beads were washed thoroughly, eluted in fresh elution buffer (1% SDS, 0.1 M NaHCO3) and de-crosslinked overnight at 65 °C. DNA samples were treated with RNase A and Proteinase K, purified with the QIAquick PCR purification kit (Qiagen), followed by qPCR analysis using the QuantiNova SYBR Green PCR Kit (Qiagen) on QuantStudio 5 Real-Time PCR Systems (Applied Biosystems) with primers described in Supplementary Information.
In vivo drug treatment
All animal experiments were approved and performed according to SingHealth Institutional Animal Care and Use Committee (IACUC) guidelines. 5–6 weeks old male NCr nude mice were purchased from InVivos (Singapore). Mice were acclimatized for 3 days before the start of the experiment. 5 × 106 WT and RR FaDu cells were resuspended in Matrigel (Corning, USA) before injecting into the flanks of NCr nude mice. Once tumors reached a minimum volume of 150mm3, mice were randomized into two groups (n = 5): Control (vehicle only), and 2.5 mg/kg panobinostat. Panobinostat was resuspended 10% DMSO + 90% corn oil, according to the manufacturer’s instructions. Treatment was administered via intraperitoneal injection (i.p.) on every alternate day for 24 or 34 days, and tumor volume and body weight were measured three times per week. Tumor volume was derived by the equation V = π/6 × L × B × H, where L is the largest superficial diameter, B is the smallest superficial diameter, and H is the height is the tumor. Tumor growth rate was calculated based on the difference between the first and the last tumor volume measured over the total number of days of treatment. All mice were sacrificed once tumors reached a maximum volume of 1500mm3 or experimental endpoint at 24 days or 34 days. Tumors were harvested for both RNA extraction and formalin-fixed paraffin embedded (FFPE) blocks for subsequent IHC analysis and TUNEL assay.
Immunohistochemistry (IHC)
Tumor tissues harvested were fixed in 10% formalin for 36 h, dehydrated, and embedded in paraffin before cutting into 5 µm sections. FFPE sections were deparaffinized and rehydrated before incubating with primary antibody, anti-Ki67 (1:3000, Cell Signaling Technology, 9449) at 4 °C overnight. HRP Rabbit/Mouse (Dako, Denmark, #K5007) was used as a secondary antibody were then incubated at room temperature for 1 h. DAB chromogen (Dako, Denmark, K5007) was used to detect Ki67-positive cells. Sections were counterstained with 10X diluted haematoxylin and mounted with Organo Limonene medium (Sigma, USA, O8015). The percentage of positively stained cells was quantified using Qupath, by using optical density to select the positive cells out of the total cells in each field.
TUNEL assay
DeadEnd™ Colorimetric TUNEL System (Promega, USA, G7130) was used to detect late apoptotic cells in the harvested tumor tissues. 5 µm FFPE sections were processed according to the manufacturer’s instructions. Sections were incubated with Terminal Deoxynucleotidyl Transferase (TdT) in a humidified chamber at 37 °C for 1 h, and then incubated with Streptavidin HRP. Apoptotic cells were stained using DAB chromogen (Dako, Denmark, K5007), counterstained with 6X diluted haematoxylin, and slides were mounted. The percentage of positively stained cells was quantified using Qupath, by using optical density to select the positive cells out of the total cells in each field.
RNA sequencing (RNAseq)
Total RNA was extracted from cells using RNeasy Mini Kit (Qiagen, USA) according to the manufacturer’s instructions. Stranded RNAseq libraries were prepared by poly(A) mRNA isolation and NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England BioLabs, USA). 150 bp paired-end sequencing was performed using Novaseq 6000 (Illumina, USA) with at least 50 million reads/samples. Adapter sequences and low-quality base calls were removed from the raw sequence reads using Trim Galore (v0.6.4) and Cutadapt (v2.10). Trimmed reads were then mapped to the hg38 reference genome using Spliced Transcripts Alignment to a Reference (STAR v2.6.1d) with standard settings. Gene quantification was performed with the “—quantMode GeneCounts” option in STAR.
Differential gene expression analysis
Differential gene expression analysis was conducted using DESeq2 v1.38.3 in R, where the equation was designed to integrate the effects of cell line types, cell phenotypes, and HDAC6 status. To define genes differentially expressed in RR relative to WT cells, we applied a threshold of log2 fold-change (FC) greater than 0.5 and an adjusted p-value smaller than 0.1 to obtain significantly differentially expressed (DE) genes. The same criteria were used to select the DE genes in RR cells with or without genetic loss of HDAC6. Interaction analysis between the DE genes identified in cell phenotypes and genetic loss of HDAC6 status was used to select for RR genes that responded to siHDAC6. This comparison was based on the dysregulation direction of the FC between two set of DE genes. Only genes showing an opposite direction of FC in both sets of DE genes were chosen as potential RR genes responding to siHDAC6.
Refinement of gene selection and geneset enrichment analysis (GSEA)
Refinement process was used to further select for genes identified from interaction analysis based on RNA abundance. This step is used to emphasize the effect of siHDAC6 in RR cells but not WT cells. Pairwise comparisons were conducted within modified and control cell types (WT: WT-control vs. WT-siHDAC6; RR: RR-control vs. RR-siHDAC6) to calculate relative changes in RNA abundance for these genes. Subsequently the ratio of relative changes between WT and RR groups were computed to quantify the magnitude of gene expression alterations. Only genes exhibiting a log2FC greater than 1 were retained, indicative of larger changes in the RR cells compared to the WT cells. The refinement process was conducted independently for each cell lines (FaDu and HK-1) and only the retained genes consensus in both cell lines were included for final selection. The final selection process was conducted to obtain the genes that related to radioresistant. GSEA was conducted using fgsea v1.24 in R to identify the enrichment pathways of these consensus genes in RR-siHDAC6 versus RR-control cells by using Gene Ontology (GO) library. The top 10 significantly positively enriched and negatively enriched pathways in siHDAC6-RR cells versus control-RR cells were selected.
Generation and validation of prognostic signature
Gene expression profile and clinical information of patients in the Genomic Data Commons (GDC) The Cancer Genome Atlas (TCGA) HNC patient cohort database were retrieved from The Broad Institute’s Firehose database, selecting only patients who received RT or adjuvant RT (n = 261), along with their mRNA abundance data. A univariate Cox proportional hazard regression model was used to assess the association of gene expression with disease free survival (DFS) of HNC patients who received RT in GDC TCGA HNC cohort. The hazard ratio (HR) from the univariate Cox regression analysis was used to identify candidate genes associated with DFS from the dataset. Following generation of a candidate gene panel, the Kaplan–Meier method was used to assess the differences in DFS time of HNC patients from low and high-expression groups, and the log-rank test was used to determine the statistical significance of observed differences between groups.
NCCS patient cohort
The National Cancer Centre Singapore (NCCS) cohort comprises 158 patients who were diagnosed with biopsy-proven NPC from August 2003 to November 2019 and received treatment with radiotherapy (RT) or concurrent chemotherapy with RT (CCRT), with or without induction chemotherapy (IC). RT was delivered using either intensity-modulated RT (IMRT) or image-guide RT (IGRT). In the definitive setting, IMRT/IGRT was delivered at doses of 74–78 Gy in 37–39 fractions, 57–60 Gy in 19–20 fractions, or 36.25–37.5 Gy in 5 fractions.
NCCS tumour sampling and RNAseq
Treatment-naïve primary tumors from the NCCS patient cohort were sampled from formalin-fixed paraffin-embedded (FFPE) diagnostic biopsies following review by an expert pathologist to identify samples with at least 70% tumor cellularity. 10 sections of 10 μM thickness were obtained, and both DNA and RNA were extracted using ReliaPrep FFPE gDNA Miniprep System (Promega) and RNAprep pure FFPE Kit (Tiangen) respectively. Stranded RNAseq libraries were prepared using TruSeq RNA Exome kit (Illumina). 150 bp paired-end sequencing was performed using Novaseq 6000 (Illumina) with at least 50 million reads/sample. Both library preparation and sequencing were carried out by NovogeneAIT Genomics Singapore Pte Ltd. Adapter sequences and low quality base calls were removed from the raw sequence reads using Trim Galore (v0.6.4) and Cutadapt (v2.10). Trimmed reads were then mapped to the hg38 human reference genome using Spliced Transcripts Alignment to a Reference (STAR v2.6.1d) with standard settings. Gene quantification was performed with the “–quantMode GeneCounts” option in STAR.
Statistical analysis
All experiments were performed in at least biological triplicates, with data presented as means ± standard deviation (SD). Student’s two-tailed t test was used for the comparison of two independent groups. A P value of < 0.05 was accepted as statistically significant. Prism 9 software (GraphPad) was used for data analysis.

Comments (0)