Cell culture
Human CRC cell lines (HCT116, HCT15, HT29, RKO, and SW480) and human colorectal normal epithelial cells (NCM460) were purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences, Shanghai Institute of Biochemistry and Cell Biology. HCT116 was cultured in McCoy’s 5a medium containing 10% fetal bovine serum and penicillin (100 U/ml)/streptomycin (0.1 mg/ml). HCT15, HT29, RKO, SW480, and NCM460 cell lines were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum and penicillin (100 U/ml)/streptomycin (0.1 mg/ml). All cell lines applied in the experiments were performed less than three passages after cell thawing.
Lentivirus preparation, plasmids and cell transfection
The shALKBH1 was constructed into pLKO.1 lentiviral vector. These plasmids were co-transfected into the 293T cells with the packaging by the standard calcium chloride transfection (lentivirus: packaging plasmid: envelope plasmid = 2:1:1). After 48 h, lentiviral supernatants were collected and concentrated by ultracentrifugation or PEG4000 + centrifugation and stored at − 80°C. The target sequence of short hairpin RNAs was as follows: shALKBH1-1, 5’-GUGAUCAAAUCUCAGCUAA-3’, shALKBH1-2, 5’-GGACA GATCTGAGCTAGAT-3’, and shNC, 5’-TTCTCCGAACGTGTCACGT-3’. Cells (HCT116 and RKO) were infected and selected with 2 mg/ml puromycin for 2 days. After selection, cells were used in the following experiments. To overexpress ALKBH1 in CRC cells, the plasmids expressing a C-terminal flag tag on the protein of ALKBH1 (ALKBH1-flag) or plasmid vectors (PV) were generated from GENEWIZ Technology. The production process of ALKBH1 enzyme activity mutant plasmids is as described in the previous article [20, 24]. In brief, a significant structural feature of hALKBH1 at the N-terminus is the Flip0 (residues 19–32). R24A/K25A/R28A/R31A in Flip0 domain completely lost demethylation activity toward 6 mA ssDNA. The arginine or lysine at positions 24, 25, 28 and 31 was mutant to alanine. Based on the overexpression plasmid of ALKBH1, the ALKBH1 mutant plasmid was constructed using the double-point mutation kit from NOVAZEN. The transfection of plasmid was performed using X-tremeGENE HP DNA Transfection Reagent according to the manufacturer’s instructions. Following treatment was administered 2 days after the transfection. Cells then were collected for further analysis.
Polyacrylamide (PA) gel manufacturing
PA-gels of varying stiffness were prepared as previously described [25]. 40% acrylamide and 2% bis-acrylamide stock solutions were mixed to prepare stiff or soft PA-gel solutions according to the protocol [26]. Ammonium persulfate (10%) and N, N, N’, N’-tetramethylethylenediamine (TEMED) were added to initiate polymerization. The gels were activated with sulfo-SANPAH (Pierce, 0.5 mg/ml in PBS) under UV light (264 nm) to cross-link extracellular matrix proteins onto the gel surface. The gels were then coated with collagen Type I (0.1 mg/ml, Corning) for 1 h at room temperature.
Human tumor samples
For immunohistochemistry (IHC) staining, we utilized a human CRC tissue microarray (HColAde075Pre-01 and OD-CT-DgCol04-003) sourced from Outdo Biotech Company (Shanghai, China). Additional human CRC tumor tissues were collected from patients undergoing endoscopy at the Endoscopy Center of Beijing Friendship Hospital, with the study being approved by the Institutional Ethics Committee of Beijing Friendship Hospital (approval number 2020-P2-290-01). Clinical information is presented in Table S1.
Animal studies
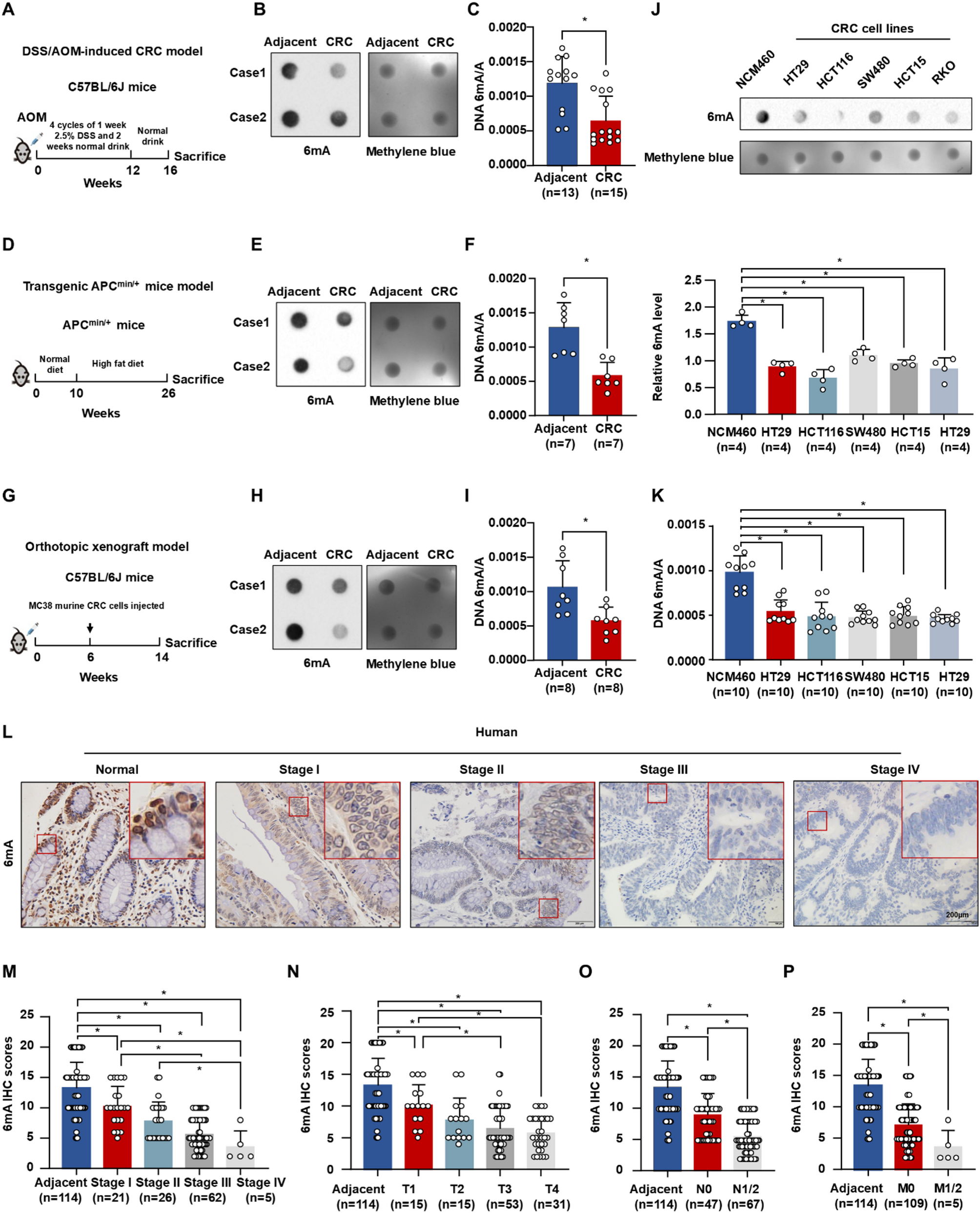
C57BL/6J-WT (aged 6–8 weeks), C57BL/6J-Apc Min/+ mice (aged 6–8 weeks) and BALB/c nude mice (aged 5–6 weeks) were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. and housed under specific-pathogen-free conditions at the Capital Medical University’s animal facility. Three CRC mice models, carcinogen azoxymethane (AOM)/ dextran sodium sulfate (DSS)-induced, transgenic ApcMin/+ and orthotopic xenograft model, were established in this study. (1) The AOM/DSS-induced CRC model, mice were earmarked and randomly assigned to groups by an independent individual. C57BL/6J mice of matching age were employed for the CRC model. Mice received an intraperitoneal (i.p.) injection of 7.4 mg/kg AOM. They underwent three cycles of 3% DSS for 7 days during 2, 5, and 8 weeks, and were sacrificed in week 16. (2) The transgenic Apc Min/+ model, ApcMin/+ mice were started on a high-fat diet at the age of 10 weeks and were sacrificed and samples were collected at 26 weeks. (3) The orthotopic xenograft model, MSI-high type CRC cell line MC38 (5 × 105 cells in 10 µL Matrigel per mouse) was orthotopically implanted into the rectum of male C57BL/6J-WT mice aged 6 weeks.
For BAPN treated mouse model, the DSS/AOM induced CRC mice and the orthotopic xenograft mice were randomly divided into BAPN-, and PBS-treatment groups. BAPN (50 mg/kg) was administered intraperitoneally every two days for four weeks in the BAPN group. For the xenograft model, approximately 2 × 10^6 HCT116 cells suspended in PBS buffer mixed with Matrigel (volume ratio 4:1) were subcutaneously injected into BALB/c nude mice. Tumor dimensions were monitored every three days, and when tumors reached a length greater than 1 cm, mice were sacrificed, and tumors were harvested for further analysis.
Antibodies, chemicals and commercial kits
Anti-6 mA antibody (202003) was purchased from Synaptic Systems. Anti-Ki67 (ab15580) antibody was purchased from Abcam. Anti-PCNA antibody (10205-2-AP), Anti-ALKBH1 antibody (27973-1-AP), Anti-N6AMT1 antibody (16211-1-AP), and Anti-GAPDH antibody (10494-1-AP) were purchased from Proteintech. Anti-METTL4 antibody (A25600) was purchased from ABclonal. Normal rabbit IgG (#2729), Anti-CDKN1A antibody (#2947), Anti-P53 antibody (#9282), Anti-p-P53 antibody (#9284), Anti-vimentin antibody (#3932) and Anti-E-Cadherin antibody (#3195) were purchased from Cell Signaling Technology. β-Aminopropionitrile (BAPN) (HY-Y1750) was purchased from MedChemExpress.
IHC staining assays
Tissues for IHC were fixed in 4% paraformaldehyde for 24 h, dehydrated through a series of alcohol solutions, and embedded in paraffin. Slides were incubated at 65℃ for 2 h, deparaffinized in xylene, and rehydrated in alcohol. Antigen retrieval was performed under high pressure, and endogenous peroxidase activity was quenched with 3% H2O2 for 10 min. Sections were blocked with goat serum for 1 h before overnight incubation at 4℃ with the primary antibody. After incubation with a secondary antibody for 1.5 h at room temperature, tissues were developed using a diaminobenzidine (DAB, ZLI-9019, ZSGB-BIO) kit and counterstained with hematoxylin. The IHC score was determined based on the intensity and extent of staining. Staining intensity was scored as 1 (no staining), 2 (weak), 3 (moderate), or 4 (strong). The extent of staining was scored as 1 (0%), 2 (1–25%), 3 (26–50%), 4 (51–75%), or 5 (76–100%). The final IHC score was calculated by multiplying the intensity and extent scores.
6 mA Dot blot and Western blot
The DNA dot blot was performed as previously reported [25]. Briefly, equal amounts of DNA were denatured with 0.4 M NaOH and 10 mM EDTA at 95℃ for 10 min, then neutralized with 2 M ammonium acetate (pH 7.0). Denatured DNA samples were spotted onto a nitrocellulose membrane and baked at 80 °C for 2 h. For western blot, equal amounts of protein were separated by 10% SDS-PAGE and transferred to polyvinylidene fluoride membranes. After blocking non-specific binding with 5% skimmed milk in TBS-T, membranes were incubated with primary antibodies diluted at 1:1000 overnight at 4℃, followed by incubation with specific secondary antibodies for 2 h at 37℃. Protein bands were visualized using Bio-Rad gel imaging system.
6 mA ELISA
Genomic DNA was extracted from human and murine CRC specimens, as well as cultured cell lines, using the QIAamp DNA Mini Kit from Qiagen. DNA 6 mA methylation levels were quantified using the Methyl Flash 6 mA DNA Methylation ELISA Kit (Colorimetric) following the manufacturer’s instructions. The methylated fraction of 0.1 ug DNA was recognized by a 6 mA antibody and quantified via an ELISA-like reaction. The percentage of 6 mA (6 mA%) was calculated from the optical density (OD) value using the provided formula. Methylated and unmethylated DNA served as positive and negative controls, respectively.
RNA isolation and quantitative RT-PCR (qRT-PCR)
Total RNA was extracted using TRIZOL and reverse transcribed as recommended. qRT-PCR was performed on Q7 real-time thermal cycler with specific primers (Table S2). mRNA expression was normalized to GAPDH and relative expression was determined using the 2−ΔΔCt method.
CCK8 assays
Approximately 2000 cells were seeded into each well of a 96-well plate. At various time points post-seeding, wells were refreshed with a medium containing 10% CCK8 substrate and incubated at 37℃ for 2 h. The absorbance at 460 nm was measured using a microplate reader, and the cell proliferation rate was calculated.
5-Ethynyl-2’-deoxyuridine (EdU) staining
Cells were incubated with 50 µM EdU at 37℃ for 2 h, followed by elution with PBS buffer. After fixation with 4% PFA, cells were treated with glycine and permeabilized with 0.5% TritonX-100. They were then incubated in 100 µL 1 × Apollo staining solution for 0.5 h in the dark. For nuclear staining, cells were treated with 1 × Hoechst 33,342 for 0.5 h in the dark, and excess Hoechst was removed with PBS buffer. Fluorescence microscopy was used to observe and photograph cell fluorescence, with ImageJ software calculating the Apollo/Hoechst positive ratio.
Colony formation assay
Transfected cells were plated at a density of 5 × 10^2 cells per well in 6-well plates and cultured at 37℃ for 14 days. Following fixation with 4% paraformaldehyde (PFA), cells were stained with 0.5% crystal violet for 30 min. Colonies were then visualized and imaged using a colony counter, and the number of colonies was quantified using ImageJ software.
Cell titer Glo (CTG) assay
Cell viability was accessed by CTG assay according to the manufacturer’s protocol. Briefly, HCT116 and RKO cells were seeded in 96 well white culture plates at a density of 1000 cells/well in 200 µl of the cell culture medium. Cells were incubated at 37˚C, 5% CO2 for 24 h. Following incubation, 100 µl of media was added to each well and treated with 100 µl of CTG reagent and kept on a shaker for 2 min. The plates were kept at room temperature for 10 min to stabilize the luminescence signal. Luminescence was measured using microtiter plate ELISA reader (Bio Tek, Winooski, Vermont, USA).
Luciferase reporter assay
Luciferase reporter plasmids containing the promoter sequences for CDKN1A and the targeting sequence of 6 mA/6 mA-Mut were transfected to measure transcriptional activation. Control groups received equivalent amounts of the pLG4 empty vector and processed identically to experimental transfection conditions. After 24 h, a Dual-Luciferase Reporter Assay System was employed to quantify firefly and renilla luciferase activities. Relative luciferase activity was calculated by normalizing firefly luciferase activity to renilla luciferase activity.
Chromatin Immunoprecipitation (ChIP)-PCR
ChIP assays were conducted using kits from Beyotime. HCT116 cells (1 × 10^7) were crosslinked with 1% formaldehyde, lysed, and genomic DNA was sheared by sonication. Anti-6 mA or anti-p53 antibodies were used to precipitate DNA fragments corresponding to 6 mA peaks. Quantitative real-time PCR was performed to determine the fold-enrichment of each fragment, with input DNA serving as a control. Primer sequences are listed in Table S2.
Stiffness measurement with nanoindentation
Tissue stiffness was measured using a ferrule-top nanoindenter setup with a PIUMA controller/drive, as previously described [25]. A probe with a 0.49 N/m spring constant and a 9 mm spherical indentation tip was used for the measurements. Specimens were fixed to slides, and the probe was brought into contact with the material surface to record load indentation and load-time data. Indentations were depth-controlled at 10 μm, with a loading and unloading period set to 2 s.
Data acquisition
RNA expression microarray data (GSE150936 and GSE117632) were sourced from the Gene Expression Omnibus (GEO) database [27, 28]. Colon adenocarcinoma (COAD) and rectum adenocarcinoma (READ) cohorts were accessed from The Cancer Genome Atlas (TCGA) via the UCSC Xena repository. Immunohistochemical staining data for ALKBH1 in normal and CRC tissues were obtained from the Human Protein Atlas (HPA) [29]. Staining intensity was extracted from the “Antibody staining” section to quantitatively assess ALKBH1 protein expression levels.
Statistical analysis
All measurements are shown as mean ± SEM from at least 3 independent biological experiments. Statistical analyses were performed using GraphPad Prism 9.0.0. Independent Student’s t-test was used to compare the difference between two groups. One-way ANOVA with Turkey’s multiple comparison was used to compare the difference among three or more groups. The 2-sided P-value < 0.05 was considered statistically significant.

Comments (0)