
Remember me
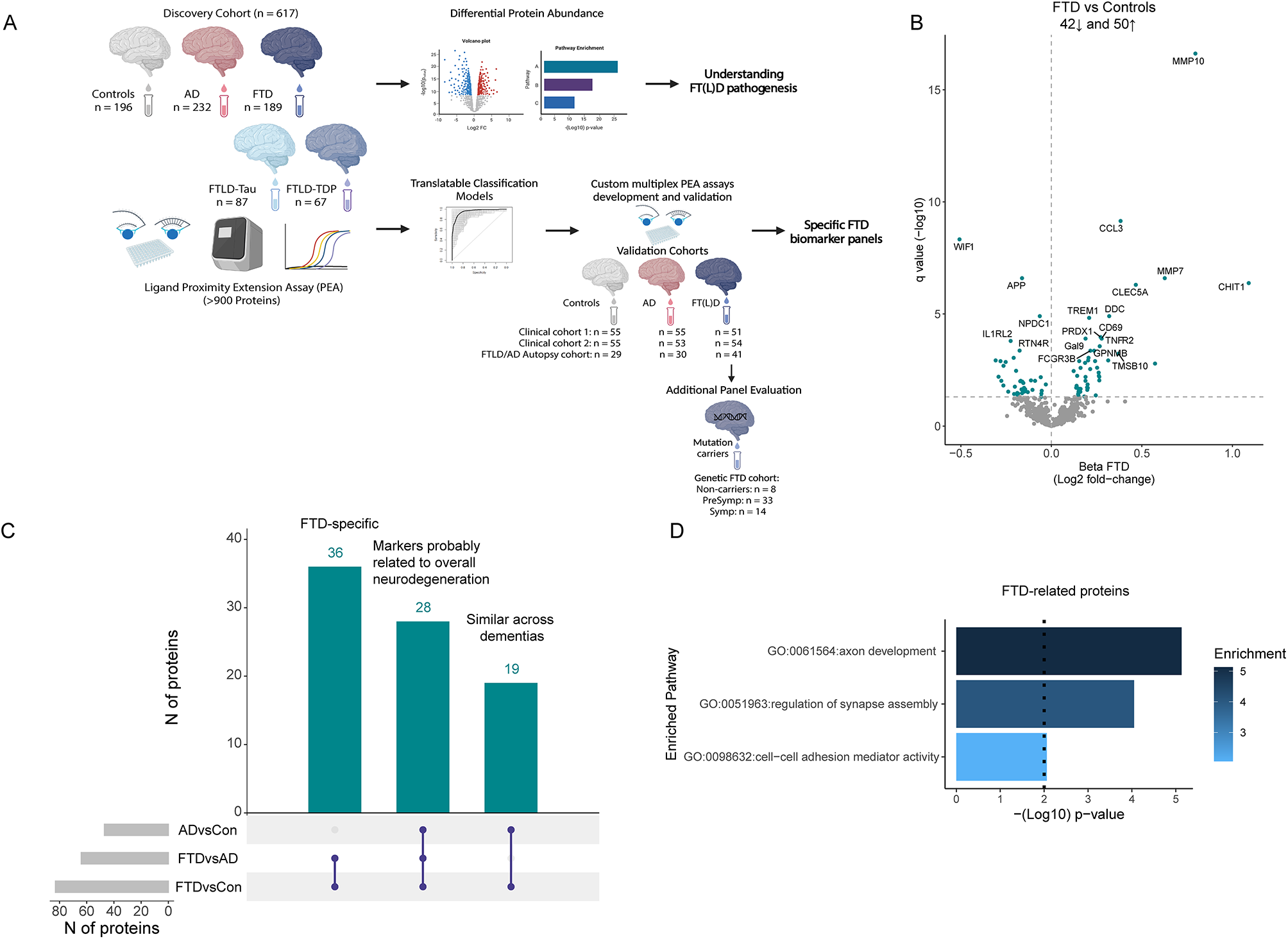
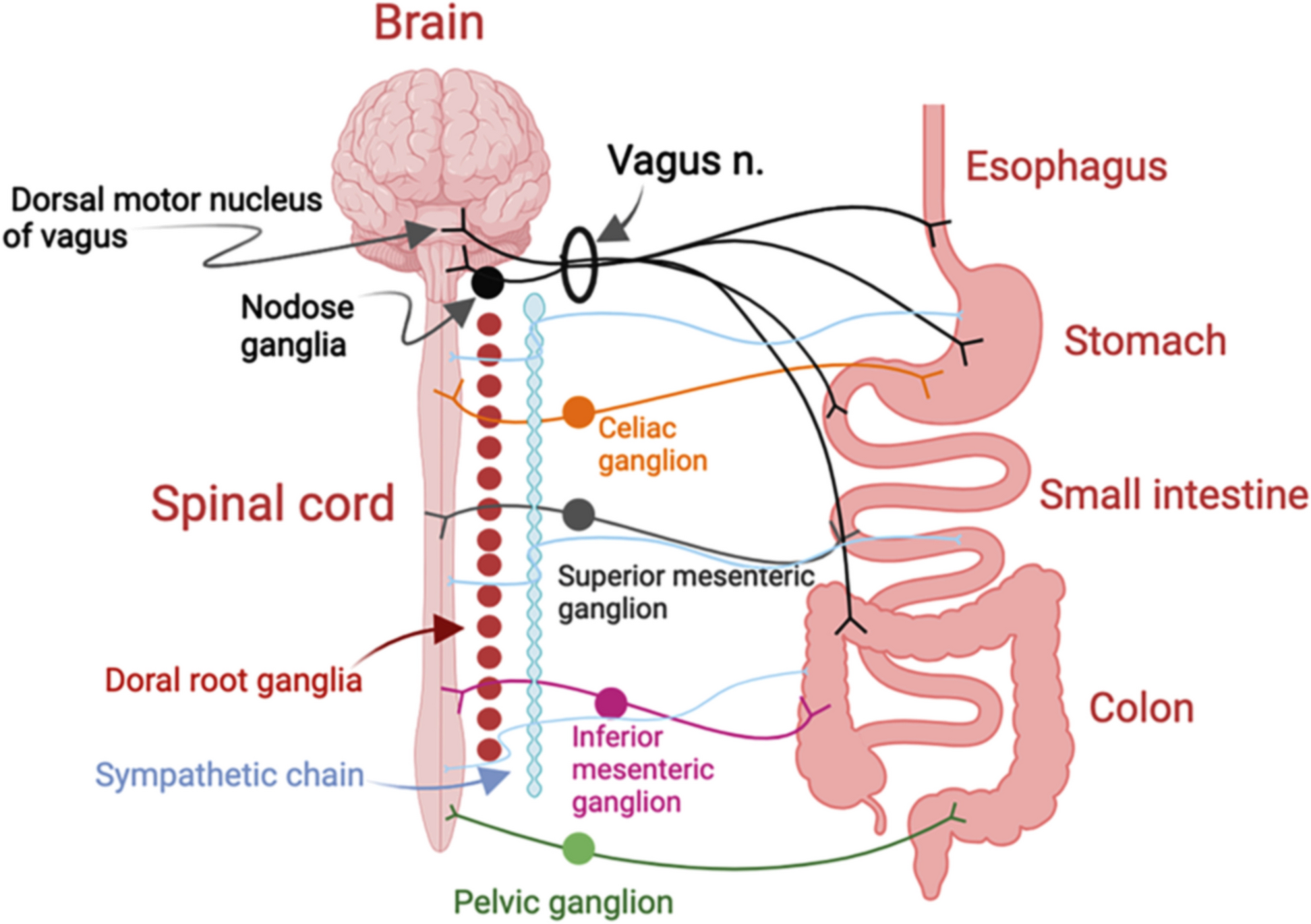
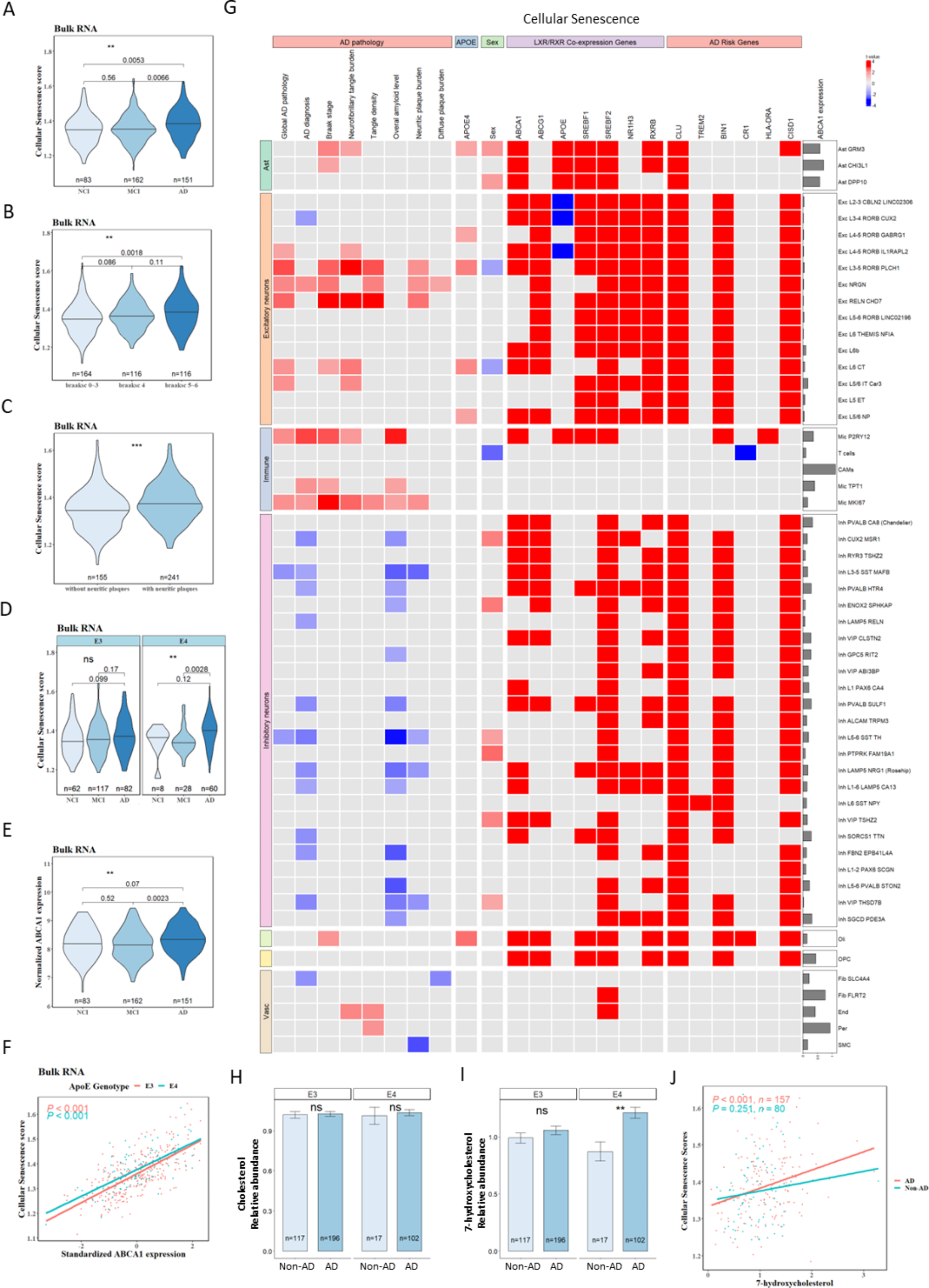
To determine whether cellular senescence is associated with ABCA1 expression in APOE4 and AD, we analyzed the databases of bulk-RNA sequencing from 632 participants and single-nucleus RNA sequencing from 427 participants, all derived from the dorsolateral prefrontal cortex (DLPFC) of postmortem human brain samples in the Religious Orders Study or the Rush Memory and Aging Project (ROSMAP). Clinical and pathological characteristics of the samples are detailed in Supplementarys Table (1) and Table (2). Validation of the RNA-sequencing findings was performed using frozen mid-frontal lobe brain tissues from 138 participants and formalin-fixed paraffin-embedded (FFPE) slides from 11 participants. To identify the proteins that regulate ABCA1 degradation, proteomics was conducted, with ABCA1 purified from HeLa cells overexpressing ABCA1 and treated with recombinant ApoE3 or ApoE4 proteins. The results from the proteomics were further validated through biochemistry and histochemistry analyses. KEGG pathway analysis of 768 shared proteins was performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) platform [32], and identified several pathways mediating ABCA1 degradation. To evaluate the impact of cholesterol reduction, 14–15 APOE4-TR mice per group were subjected to behavioral tests and treatments, with testers blinded to group allocation. All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Southern California, ensuring minimal animal stress and use. For all experiments, the number of replicates, statistical tests, and P values are reported in the figure legends.
Analysis of bulk-RNA sequencing dataDatabaseFiltered raw counts were downloaded from the Synapse AD Knowledge Portal (https://www.synapse.org/#!Synapse:syn9702085) with Synapse ID: syn8456637.
Source data were collected from a cohort of 632 subjects from ROSMAP [33]. Both studies were approved by the Institutional Review Board of Rush University Medical Center. All participants signed informed and repository consent forms and the Anatomical Gift Act. Based on clinical diagnosis at the time of death, 83 study participants were diagnosed with no cognitive impairment (NCI), 162 with mild cognitive impairment (MCI), and 151 with AD dementia [34,35,36]. CERAD Braak staging represents the amount and distribution of neuritic plaques and neurofibrillary tangle pathology [37, 38]. APOE genotype was determined as previously described [39]. Detailed information regarding all samples is provided in Supplementary Table 1.
Gene set enrichment analysis (GSEA)The R package GSVA (method option “ssGSEA”) was used to calculate the pathway enrichment score of each subject. The annotated gene sets were retrieved from MsigDB. REACTOME_CELLULAR_SENESCENCE (M27188) was selected to represent cellular senescence. The pathway score was tested to determine the differences between the variables of interest.
Analysis of single nucleus-RNA sequencing dataDatabasePost-QC counts and ROSMAP metadata were downloaded from the Synapse AD Knowledge Portal (https://www.synapse.org/#!Synapse:syn52293417) with Synapse ID: syn2580853. Source data were collected from a sample of 427 subjects from ROSMAP [40]. Detailed information about the samples is provided in Supplementary Table 2. Nuclei were isolated from frozen postmortem brain tissues and subjected to droplet-based single-nucleus RNA sequencing (snRNA-seq).
Gene set enrichment analysis (GSEA)The R package GSVA (method option “ssGSEA”) was used to calculate the pathway enrichment score of each cell. Single sample GSEA (ssGSEA) is a non-parametric method that calculates the normalized difference in empirical cumulative distribution functions (CDFs) of gene expression ranks inside and outside the gene set, representing the enrichment score for the gene set. The annotated gene sets were retrieved from MsigDB. REACTOME_CELLULAR_SENESCENCE (M27188) and REACTOME_SENESCENCE_ASSOCIATED_SECRETORY_PHENOTYPE_SASP (M27187) were selected to represent cellular senescence and SASP. The linear mixed effect model was used to test if the pathway score was different between the variables of interest, using the formula: ~ size factor (i.e. the library size of each cell) + interested variables + (1|projid (i.e. sample id)).
SenTraGor (STG) stainingThe formalin-fixed paraffin-embedded (PPFE) human brain slides containing middle frontal lobe region (ROS) were deparaffinized following the protocol (xylene, 3 min each time for two times; xylene 1:1 with 100% ethanol, 3 min each time for two times; 100% ethanol, 3 min each time for two times; 95% ethanol, 3 min each time for two times; 70% ethanol, 3 min each time for two times; 50% ethanol, 3 min each time for two times; running cold tap water to rinse for 5 min). The slides were then subjected to antigen retrieval using a Sodium Citrate buffer (10 mM Sodium Citrate, 0.05% Tween 20, pH 6.0) for 30 min. The slides were washed once with TBS for 5 min and incubated with STG (Bio-Techne, #7555, 2 mM, dissolved in 100% ethanol, filtered with 0.45 µM filter) for 10 min at room temperature (RT). After incubation, the slides were washed twice with 50% ethanol followed by one time wash with TBST (0.025% Triton X-100 in TBS) for 5 min each wash. Then, primary antibodies (goat anti-biotin and anti-GFAP) were added to the slides and incubated overnight at 4 °C. Negative control was set as without goat anti-biotin antibody or anti-GFAP antibody. After washing three times with TBST, the slides were incubated with fluorescein-conjugated secondary antibodies at room temperature for 1 h. After washing three times with TBST, the slides were incubated with an autofluorescence quencher at room temperature for 5 min. After washing thrice with TBST, the slides were mounted with mounting medium (F4680; Sigma). Images were acquired using Leica SP8 confocal microscope (63x objective) and analyzed using the ImageJ software (NIH). The slide order number was used in image acquisition and analysis to blind the disease groups or genotypes.
Human brain tissue homogenate preparationFrozen mid-frontal lobes of postmortem human brains were obtained from the Rush Alzheimer’s Disease Center (RADC) at the Rush University Medical Center. The Religious Orders Study (ROS) was approved by the Institutional Review Board (IRB) of Rush University Medical Center [41]. Detailed information on these samples is presented in Supplementary Table 3.
To extract soluble and insoluble proteins, frozen middle frontal lobe brain tissues were weighed and homogenized with TBS (1:15, w/v) containing a protease inhibitor cocktail and phosphatase inhibitor cocktail. The homogenate was centrifuged at 15,000 x g for 1 h at 4 °C. The supernatant was collected as the TBS fraction, and the pellets were incubated with TBSX (1% Triton X-100) (same volume as TBS) overnight with agitation at 4 °C. The next day, the homogenate was centrifuged at 15, 000 × g for 1 h at 4 °C and the supernatant was collected as the TBSX fraction. The protein concentrations of all fractions were measured using a bicinchoninic acid kit and used for further experiments. For ABCA1 detection, TBSX fraction samples were added to 4x Laemmli sample buffer (without boiling) and subjected to 12% SDS-PAGE for western blotting.
Lysosome enrichment with LAMP-2 immunoprecipitationLAMP-2 antibody (1.2 µg) was added to the TBSX fraction samples (300µL) of AD E3/3 and AD E3/4 individuals. After incubation for 1 h at 4 °C, 20 µL Dynabeads Protein A/G (88802; Thermo Scientific) was added and incubated with rotation overnight at 4 °C. After two washes with 1 mL of ice-cold 0.1% TBST, 20 µL of elution buffer (50 mM Glycine, pH2.8) was added and incubated for 2 min at room temperature. The elution was quickly neutralized with 2 µL of Tris buffer (1 M, pH 8.5). Then LAMP-2 enriched samples were added to 4x Laemmli sample buffer (without boiling) and loaded onto a 7.5% SDS-PAGE gel for western blotting.
ABCA1 complex purificationHeLa cells expressing ABCA1-GFP were cultured in DMEM (Corning, 10–013) with 10% fetal bovine serum (FBS) (Omega Scientific, FB-12) and 1% antibiotic-antimycotic (anti-anti) (Thermo Fisher, 15240062) at 37 °C in a 5% CO2 incubator. When the cells were ready, recombinant APOE3 or APOE4 (0.2 µM) was added for 4 h. After treatment, the cells were scratched, collected with DPBS (Corning, 21-031-CV), and washed twice with ice-cold DPBS. Then, 5 volumes of lysis buffer (0.5% NP40, 350 mM Nacl, 20 mM Hepes, 1.2% Triton X-100, complete cocktail) were added and incubated with rotation for 5 min at 4 °C. The cell lysate was centrifuged for 15 min at 13,000 rpm and 4 °C, and the supernatants were collected for immunoprecipitation.
100 µL of GFP-Trap Agarose (gta-20, Chromotek) was first washed with IP buffer (PBS 7.5–7.9; 200 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 1 mM DTT and 0.2 mM PMSF) three times and then added to 2 mL supernatant of HeLa (ABCA1-GFP) cell lysates and incubated with rotation overnight at 4 °C. The next day, agarose was washed with IP buffer three times with rotation at 4 °C. After washing, a 2-fold volume of glycine (0.1 M, pH 2.7) was used to elute the ABCA1 complex and neutralized with 1/10 volume of Tris-HCl (1 M, pH 9.5) after elution. Parts of elution were used for SDS-PAGE analysis, and other parts were added with TCA (SA433-500, Fisher Scientific) to a final concentration of 20% and incubated for 15 min to precipitate proteins. After centrifugation (13,000 rpm, 4 °C, 30 min), the pellet was collected and washed once with 1mL of acetone (A949-1, Fisher Scientific). After removing the acetone, the protein pellets were airdry for 3–4 min at room temperature to ensure that no liquid remained. Finally, the protein pellet was sent to a mass spectrometry facility (Harvard Center for Mass Spectrometry).
For ABCA1 degradation experiments, Hela cells expressing ABCA1-GFP were treated with recombinant APOE3 or APOE4 (0.2 µM) (Academy Biomedical Company) for different time points. After treatment, cells were lysed with radioimmunoprecipitation assay (RIPA) buffer (9806, Cell Signaling Technology, CST), and total ABCA1 protein levels were detected by western blotting.
Analysis of mass spectrometry dataNormalized Spectral Abundance Factors (NSAFs) [42] were used to measure the relative abundance and calculate the fold change for each protein. To obtain the NSAF value for a specific protein, the spectral abundance factor (SAF) value was first calculated by dividing the spectral count of the protein by its amino acid length. The NSAF was then calculated by normalizing the SAF value to the total SAF values of all the identified proteins within each sample. This approach allows for the accurate assessment of protein abundance across different samples.
ImmunocytochemistryImmortalized astrocytes were grown in 8-well cell culture chamber slides (Bioland Scientific, 07-2108). After fixing with 4% paraformaldehyde for 15 min, the cells were permeabilized with 1% Triton X-100 for 30 min and then blocked with 10% goat serum and 0.1% Triton X-100 for 1 h at room temperature. After blocking, the cells were incubated with anti-ABCA1 antibody (Abcam, ab18180) (1:100) and anti-Caveolin-1 antibody (CST, 3267 S) (1:200) or anti-AP2B1 antibody (GeneTex, GTX79316) (1:200) overnight at 4 °C. After washing, the cells were incubated with Alexa Fluor Plus 488 or 594 labelled Goat anti-Mouse IgG (Thermo Fisher, A32723/A-11005, 1:200) and Alexa Fluor 594 or 488 labelled Donkey anti-Rabbit IgG (Thermo Fisher, A21207/A-21206) (1:200) for 1 h at room temperature in the dark. After washing, the nuclei were stained with Hoechst 33,258 (Thermo Fisher, H3569) (1:12,000) for 5 min. After washing, the slides were mounted with mounting medium (F4680; Sigma). Images were captured using a Zeiss Axiovert 200 M Inverted Fluorescence Microscope (20x objective). Images were analyzed using ImageJ software (NIH).
Cell culturePrimary astrocytes were obtained from APOE3-TR and APOE4-TR mouse pups and cultured as previously described [43]. Briefly, cerebral cortices from 1 to 3-day-old neonatal mice were dissected in ice-cold Hanks’ balanced salt solution (HBSS) (Corning, 21-021-CV) and digested with 0.25% trypsin for 20 min at 37 °C. Trypsinization was stopped by adding a 2-fold volume of DMEM (Corning, 10–013) with 10% fetal bovine serum (FBS) (Omega Scientific, FB-12) and 1% antibiotic-antimycotic (anti-anti) (Thermo Fisher, 15240062). The cells were dispersed to a single-cell level by repeated pipetting and filtered through 100 μm cell strainers (VWR, 10199-658). After filtering, the cells were centrifuged for 5 min at 1000 rpm and resuspended in culture medium (DMEM, Corning, 10–013) supplemented with 10% FBS and antibiotics. The cells were then seeded in a 75 cm2 flask and cultured at 37 °C in 5% CO2. The medium was changed the next day and replaced every three days. The mixed glial cultures reached confluence after 7–10 days. The cells were then shaken at 250 rpm for 16 h at 37 °C to remove the microglia and oligodendrocyte progenitor cells. The remaining cells were harvested by trypsin digestion. At this stage, the culture contained 95% astrocytes, which were used for further experiments.
Immortalized mouse astrocytes derived from human APOE3 and APOE4 knock-in mice were gifts from Dr. David Holtzman and grown in DMEM/F12 (Corning, MT10090CV) containing 10% FBS, 1mM sodium pyruvate (Thermo Fisher, 11360070), 1 mM geneticin (Thermo Fisher, 10131-035) and 1% anti-anti.
Cell plasma membrane protein preparationTo study cell plasma membrane proteins, biotinylation of cell surface proteins was performed to isolated cell plasma membrane protein. Briefly, cells were washed twice with cold PBS followed by incubation with 0.5 mg/ml sulfo-NHS-SS-biotin (Thermo Fisher Scientific, PG82077) in PBS for 30 min at 4 °C with shaking. The reaction was quenched by rinsing cells with 50 mM glycine in PBS. The cells were then lysed with RIPA buffer (CST, 9806) containing a protease inhibitor cocktail, followed by centrifugation at 12,000 × g for 10 min at 4 °C. The supernatant was collected, and protein concentrations were measured using a BCA kit (Thermo Fisher, 23225). The radioimmunoprecipitation assay (RIPA) fraction represented the total protein fraction. Then, 100 µg of the RIPA fraction from each sample was incubated with 40µl of NeutrAvidin agarose (Thermo Fisher Scientific, 29200) for 2 h at 4 °C. The agarose was then washed three times with PBS containing a protease inhibitor cocktail, followed by boiling with 100 µL 2x sample buffer for 5 min at 95 °C. After centrifugation (2,500 x g, 2 min, room temperature), the supernatants were collected as plasma membrane proteins. Membrane and total protein analyses were performed using western blotting.
ImmunoprecipitationHeLa (negative) or HeLa (ABCA1-GFP) cells were lysed in RIPA buffer containing protease and phosphatase inhibitors. 10 µL of GFP-Trap Agarose was added to 200 µL cell lysates (1 µg/µL) and incubated with rotation overnight at 4 °C. The next day, agarose was washed with IP buffer three times with rotation at 4 °C. After washing, a 2-fold volume of diluted (2x) sample buffer (Bio-Rad, 1610747) was added and boiled for 5 min at 95 °C. After centrifugation (1,500 rpm, 5 min, room temperature), supernatants were collected for SDS-PAGE and western blotting.
Astrocytes were lysed in radioimmunoprecipitation assay (RIPA) buffer containing protease and phosphatase inhibitors. 1.5 µg antibody (anti-ABCA1) was added to 400 µL cell lysates (1 µg/µL) and incubated with rotation for 1 h at 4 °C. Then, 25 µL Dynabeads Protein G (Thermo Scientific, 10004D) was added and incubated with rotation overnight at 4 °C. After three washes with 1 mL of ice-cold 0.1% TBST, 25 µL of diluted (1.5 ×) sample buffer was added to the beads and boiled for 5 min at 95 °C. The supernatants were collected and subjected to SDS-PAGE and western blotting.
Western blottingThe cell lysates, brain homogenates, or immunoprecipitation protein complexes were separated on 4–15% mini-precast protein gels (Bio-Rad, 4561086) under reducing conditions and then transferred onto nitrocellulose membranes (Bio-Rad, 1704270). After transfer, the membranes were blocked with 5% fat-free milk (Bio-Rad, 1706404) in TBST for 1 h at room temperature, followed by overnight incubation with the primary antibody in 5% BSA at 4 °C. The membranes were then incubated with an HRP-conjugated secondary antibody for 1 h at room temperature. A chemiluminescent HRP substrate (Millipore, WBKLS0500) was used for detection. The Fujifilm LAS-4000 imager system or X-ray film was used to capture images, and densitometric quantification was performed using Gel Quant NET software. The antibodies used are listed in Supplementary Table 5.
siRNA and plasmid transfectionsNon-targeted (NT), Caveolin-1 and AP2B1 siRNAs were obtained from Dharmacon. Cells were seeded in 60 mm dishes or 24-well plates and cultured overnight. The siRNAs (10–20 nM) were transfected using the jetPRIME reagent (Polyplus Transfection, catalog #114). Protein expression was detected using western blotting 48 h after transfection.
The mouse Caveolin-1 (Myc-DDK-tagged) expression plasmid (MR201562) was purchased from Origene and purified using an endo-free plasmid DNA midi kit (Bioland Scientific, PD03-21). The pCMV-GFP expression plasmid was used as the control. The cells were seeded in 24-well plates and cultured overnight. The plasmid (0.125 µg) was transfected using the jetPRIME reagent. Protein expression was detected using western blotting 48 h after transfection.
Cholesterol effluxImmortalized astrocytes were seeded in 24-well plate (0.12 × 105 cells/500 µL) and cultured overnight. The cells were transfected with 20nM siRNA for 24 h, followed by labeling with 1µCi/mL (3 H) cholesterol (Moravek, MT9112) using serum-free DMEM/F12 containing 2 mg/ml fatty acid-free BSA (Sigma-Aldrich, catalog #A9647), and 2 µg/ml acyl-coenzymeA: cholesterol acyltransferase inhibitor SANDOZ (Sigma-Aldrich, catalog #S9318) for 24 h. After washing once with serum-free culture medium, the cells were treated with recombinant CS-6253 peptide (1 µM in DMEM/F12 containing 2 mg/mL fatty acid-free BSA, 2 µg/mL SANDOZ, and 200 µL/well) for 4 h. After treatment, the cell culture medium was collected and transferred to scintillation vials filled with 3 mL of the scintillation mixture. The cells were solubilized in 0.5 N NaOH (200 µL), neutralized with PBS, and then transferred to scintillation vials filled with 3 mL of scintillation mixture. After vigorous mixing, vials were counted using a Beckman LS6500 liquid scintillation counter (Beckman Coulter). The efflux of cholesterol was assessed by the ratio of cholesterol in the medium to the total cholesterol (medium and cell lysate).
Cholesterol loading and depletionImmortalized astrocytes or baby hamster kidney (BHK) cells were seeded in 24-well plates (0.5 × 105 cells/500 µL) and cultured overnight. Cells were washed twice with serum-free medium, and human LDL (10 µg/mL diluted in serum-free medium containing 2 mg/mL fatty acid-free BSA) (Sigma, LP2-2MG) was added for 24 h. The cholesterol levels in the cells were determined using a cholesterol assay kit (filipin III staining, Abcam, ab133116). Cellular caveolin 1 protein levels were determined by western blotting. Human iPSC astrocytes were seeded in 48-well plates (0.5 × 105 cell/300µL) and cultured for 48 h. Cells were first loaded with LDL as described above and treated with 1 mM methyl-β-cyclodextrin (MβCD) for 2 h. Cellular cholesterol levels were determined using filipin staining. For parallel experiments, after MβCD treatment, cells were stimulated with TNFα (100 ng/mL) and IFNγ (100 ng/mL) for 16 h. RNA was extracted from each condition using an RNA extraction kit (Thermo Fisher, K0731).
ABCA1 knock down cell line and cholesterol loadingAn ABCA1 knockdown astrocyte cell line was created by transfection with ABCA1 CRISPR Gene Knockout Kit (ABCA1-sgRNAs and Cas9 recombinant protein, Synthego). Three days after transfection, the single cell was seeded and cultured for two weeks. ABCA1 knockout or knockdown clones were validated by western blotting using ABCA1 antibody. ABCA1 WT or ABCA1 knockdown astrocytes were seeded in 24-well plates (0.5 × 105 cell/500 µL) and cultured overnight. Cells were treated with methyl-β-cyclodextrin (MβCD, 5 mg/mL) for 2 h and then loaded with LDL (100 µg/mL) for 2 h. Then, the cells were lysed for further analysis.
ABCA1 induction and cholesterol loading in BHK cellsBHK cells were seeded in 24-well plates (0.5 × 105 cell/500 µL) and cultured overnight. ABCA1 expression was induced with Mifepristone (0.1 nM) treatment for 16 h. Then, cells with or without ABCA1 were loaded with LDL (100 µg/mL) for 24 h. The cells were lysed for further analysis.
Animals and treatmentAll animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Southern California. Animals were housed under a standard 12-hour light/dark cycle with water and chow diet in a pathogen-free animal facility at the University of Southern California. Every effort was made to reduce animal stress and minimize animal use. ABCA1fl/fl mice were obtained from Dr. John Parks of Wake Forest University School of Medicine. Nestin-cre mice (Strain #:003771) were purchased from Jax. ABCA1 conditional knockout mice were generated by crossing ABCA1fl/fl mice with nestin-Cre mice. ABCA1 genotype was confirmed by PCR genotyping. Ten-month-old wild type mice and ABCA1 conditional knockout mice were euthanized with CO2 and perfused with cold PBS. Brains were collected for further experiments.
The APOE3-TR and APOE4-TR mice were purchased from JAX. APOE3 and APOE4-TR mice of different ages were euthanized using CO2 and perfused with cold PBS. Brains were collected for further experiments.
APOE4-TR male and female mice (8 months old) were randomized to receive subcutaneous injections of 2 g/kg body weight 2-hydroxypropyl-β-cyclodextrin (HPCD) (Sigma, 332607-25G) in phosphate-buffered saline (PBS) or PBS twice a week for eight weeks. The mice were then subjected to behavioral tests, and the testers were blinded to the treatment allocation. The injection was continued during behavioral tests (two weeks). Finally, after collection of the cerebrospinal fluid (CSF) and plasma, the mice were anesthetized and subjected to cardiac perfusion with ice-cold PBS. The brains were dissected, and one hemisphere was post-fixed in 4% paraformaldehyde (PFA) for cryostat sectioning, while the other was used for biochemistry assays and RNA purification.
Novel object recognition (NOR)This was performed as previously reported with some modifications [44]. On day 1, mice were acclimated to the test room for 1 h followed by free exploration for 5 min in the test arena (without objects). On day 2, mice were placed into the same arena containing two identical objects and allowed to freely investigate both for 5 min (training trial). After an interval of two hours, the mice were placed back in the chamber with one novel object and one familiar object and allowed to explore for 5 min (testing trial). After each trial, the testing area and objects were thoroughly cleaned with 70% ethanol solution. The training and testing trials were recorded with a high-resolution camera, and the number of touches to both objects of each mouse in the testing trial was analyzed using the EthoVision XT software (Noldus). Mice touching the object with the nose were identified as one instance of nose touch. The movement distance, total touch times for both objects, and ratio of nose touch times of the novel object to the old object were calculated. One mouse in the HPCD group was excluded because of a fighting wound.
Cell lysate and mouse brain homogenate preparationImmortalized or primary astrocytes were lysed with 1x RIPA buffer (CST, 9806) containing protease inhibitor cocktail (Sigma, P8340) and phosphatase inhibitor cocktail (Sigma, P0044), followed by centrifugation at 15,000 × g for 10 min at 4 °C. The supernatant was then collected for further analysis. The mouse cerebral cortex was weighed, and RIPA buffer containing a protease inhibitor cocktail and phosphatase inhibitor cocktail was added at a ratio of 1:30 (w/v). The tissue was then homogenized using a 2 mL glass Dounce tissue grinder, followed by centrifugation at 15, 000 × g for 1 h at 4 °C. The supernatant was collected, and the concentration was measured using a BCA kit.
To extract soluble and insoluble proteins, mouse and human brain samples were weighed and homogenized with TBS (1:15, w/v) containing protease and phosphatase inhibitor cocktails. The homogenate was centrifuged at 15,000 x g for 1 h at 4 °C. The supernatant was collected as the TBS fraction, and the pellets were incubated with TBSX (1% Triton X-100) (same volume as TBS) overnight with agitation. After centrifugation (15,000 × g, 1 h at 4 °C), the supernatant was collected as the TBSX fraction. The pellets were dissolved in 150–200 µL guanidinium chloride (GnHCl) (5 M, pH 7.5) and incubated overnight at room temperature with agitation. After centrifugation (15,000 × g, 1 h at 4 °C), the supernatant was collected and dialyzed with a dialysis device (MINI Dialysis Device, 3.5 K MWCO, 0.1 mL, Thermo Scientific) in TBS overnight at 4 °C. The solution was collected as the GnHCl fraction. The protein concentrations of all fractions were measured using a bicinchoninic acid kit and used for further experiments.
Immunohistochemistry and immunofluorescence staining of mouse brain slidesFixed mouse brains were sectioned using a Leica cryostat at a thickness of 10 μm. Slides were equilibrated for 30 min at room temperature and rinsed twice with PBS. After blocking with blocking buffer (10% goat serum + 1% BSA + 0.2% Triton X-100 in PBS) at room temperature for 1 h, the sections were incubated with diluted primary antibodies (GFAP, Iba-1) and incubated overnight at 4 °C. After washing, the slides were incubated in a 0.3% H2O2 solution in PBS at room temperature for 10 min to block endogenous peroxidase activity. Then, the SignalStain® Boost IHC Detection Reagent (#8114, Cell Signaling Technology) was added to the slides and incubated at room temperature for 1 h. After washing, DAB substrate solution was added to the slides and developed for 2–5 min until the desired color intensity was reached. After washing, the slides were mounted with mounting medium (F4680; Sigma). Images were taken using an Olympus Microscope (10x objective). Images were analyzed using the ImageJ software (NIH).
For ABCA1 and LAMP1 double staining, after overnight incubation with the primary antibody (ABCA1, Abcam, ab18180; LAMP1, CST, #99437), the fluorescein labelled secondary antibody was added to the slides and incubated at room temperature for 1 h (protected from light). After washing, the slides were mounted with mounting medium with DAPI (Sigma, F6057). Images were taken using Leica SP8 confocal microscope (63x objective) or a slide scanner (Z1, 20x objective, Zeiss Axio Scan, Oberkochen, Germany) and analyzed using the ImageJ software (NIH). For p21 and pH2A.X staining, after overnight incubation with the primary antibody (p21, CST, #37543; pH2A.X, CST, #9718), the ABC kit (Vector Laboratory, PK-4001) and Cyanine 3 tyramide (Bio-techne, #6457) were performed to obtain the fluorescence signals. After washing, the slides were mounted with mounting medium with DAPI. Images were taken using Leica SP8 confocal microscope (63x objective) and analyzed using the ImageJ software (NIH). The slide order number was used in image acquisition and analysis to blind the groups or genotypes.
Lysosome isolationThirty to fifty mg of brain tissues were used to isolate lysosomes with the lysosome isolation kit (Minute Lysosome Isolation Kit, Invent Biotechnologies, LY-034) following the manuals. Briefly, the tissues were homogenate with a plastic rod for 1 min, followed by the centrifugation at 16,000 x g for 30 s. The filter was discarded, and the pellet was resuspended by vigorously vertexing for 10 s, followed by centrifugation at 2,000 x g for 3 min. The supernatant was collected to a fresh 1.5 mL microfuge tube and centrifuged at 4 °C for 15 min at 9,000 x g. After centrifugation, the supernatant was carefully transfer to a fresh 1.5 mL tube and spined at 16,000 x g at 4 °C for 30 min. After centrifugation, the supernatant was removed completely. Then, the pellet was resuspended in 200 µL of cold buffer A by pipetting up and down 60–100 times and vortex vigorously for 20 s, followed by centrifugation at 2,000 x g for 4 min. The supernatant was transferred to a fresh 1.5 mL tube and added 100 µL of buffer B, and vortexed briefly vortex to mix well (the supernatant to buffer B ratio is 2:1). The mixture was incubated on ice for 30 min and centrifuged at 11,000 x g for 10 min. After removing all the supernatant, the pellet (lysosomes) was resuspended in 50 µL of RIPA buffer for further analysis.
qPCRThe cells and brain specimens were harvested, and RNA was extracted using an RNA extraction kit (Thermo Fisher, K0731). cDNA synthesis was performed using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher, 4368814). qPCR was performed using the 2x SYBR Green I-based qPCR master mix (Bioland Scientific, QP01-00) on Siemens VERSANT kPCR Molecular System. The primers were listed in Supplementary Table 6. All primers were synthesized by Integrated DNA Technologies. The Ct value of the targets was normalized to 18sRNA.
Isolation of cell types from mouse brainCell types isolation from mouse brain was performed following the published protocol [45]. Briefly, mice were euthanized with CO2 and perfused with PBS. The brains were harvest and cerebra were collected. Then, the cerebra were dissociated in the enzyme digestion mix (collagenase A (Sigma, 10103586001) + DNase I (Sigma, 10104159001) by passing three separate flame-polished Pasteur pipets to a single cell suspension. After centrifugation, the debris/myelin was removed by percoll (Sigma, GE17-0891-02) gradient centrifugation. The myelin fraction and total cells mixture were collected separately. Microglia from total cells mixture were purified by incubating with CD11b microbeads (Miltenyi Biotec,130-093-636) and passing LS magnetic column (Miltenyi Biotec, 130-042-401). CD11b negative population were also collected. Myelin was separated using gradient centrifugation. After collection, the different cell types were lysed with RIPA for western blotting detection.
Ion-mobility analysisHDL particles in mouse CSF and plasma were determined by ion mobility analysis as previously reported [46]. Samples treated with dextran sulfate were introduced into a charge-reducing electrospray (TSI Inc., model 3482) every 13 min by automated loop injections using an integrated autosampler (Teledyne CETAC Technologies, model MVX-7100). The electrospray settings were as follows: voltage 2.0 kV, CO2 flow 0.15 slmp, and airflow 1.5 slmp. The differential mobility analyzer (TSI Inc., model 3085), coupled to a condensation particle counter (TSI Inc, model 3788), scanned particles 4.45 to 63.8 nm for 180 s. The generated data were analyzed using Fityk (version 1.3.1), as previously described, and graphed using OriginPro software (version 2021). Voigt probability distribution curves were generated from particle count (#/mL) vs. diameter range for lipoprotein subclasses and normalized by dividing subclasses by the sum of peak areas from all lipoproteins present within the spectrum.
Oxysterol measurement by LC-MS/MSThe Oxysterol Derivatization MaxSpec Kit and oxysterol standards were obtained from Cayman Chemical Company. Brain tissues were weighed and homogenized with steel beads in ethanol. After centrifugation at 10,000 x g for 10 min at 4 °C, 800 µL of supernatant was transferred to fresh tubes and diluted with water to a final concentration 20% ethanol. The diluted samples containing oxysterols were then extracted using Oasis HLB SPE cartridges (60 mg) that were conditioned with methanol (1 mL) and water (1 mL). Oxysterols were eluted using 70% ethanol (1 mL) and eluted again using the same procedure where the combined samples were evaporated to dryness using a steady stream of N2 gas. The dried oxysterol fractions were derivatized with Girard’s reagent P according to manufacturer’s instruction using an Oxysterol Derivatization MaxSpec Kit (Cayman Chemical, #601540). 5 µL was injected to an Agilent 1100 HPLC linked to API4000 mass spectrometer (Sciex). A Poroshell 120 EC-C18 column (2.7 μm, 4.6 × 100 mm, Agilent) was used for chromatographic separation. The mobile phase consisted of water + 0.1% formic acid (A) and methanol + 0.1% formic acid (B) with the following gradient: 0 min 50% B; 7 min 98% B; 16 min 98% B; 16.1 min 50%B; 20 min 50%B. Flow rate was set at 500 µL/min and column temperature was maintained at 40 °C. The MS was operated in positive ESI mode and the target analytes were quantified using their optimized MRM transition. Peak integrations were manually reviewed using Skyline software (v. 23.1, MacCoss Lab) and peak areas were normalized by tissue weight. The internal standard, 7β-hydroxy Cholesterol-d7, was used a corrective factor for sample processing and recovery. The corresponding GP-d5 analytes were used as a corrective factor to account for variance in injection volume and ionization efficiency for each sample and to confirm analyte retention times.
Generation of human iPSC-astrocytesAPOE isogenic human iPSC–derived neural progenitor cells (NPC) were generated in the TCW laboratory [47]. Dissociated forebrain NPCs were differentiated into astrocytes in astrocyte medium (#1801, ScienCell), as previously described [48]. Briefly, forebrain NPCs were maintained at a high density on poly L-rrnithine hydrobromide (#P3655-50MG, Sigma) and laminin (#23017015, Thermo Fisher)-coated plates and cultured in NPC medium [DMEM/F12 (Corning, MT10090CV), 1 x N2 supplement (Thermo Fisher, 17502048), 1xB27 supplement (Thermo Fisher, 12587010), 1 mg/mL laminin, and 20 ng/mL FGF2 (Thermo Fisher 13256029)]. The cells were split at approximately 1:3 to 1:4 every week with Accutase (Sigma, SCR005). NPCs were differentiated into astrocytes by seeding dissociated single cells at a density of 15, 000 cells/cm2 on Matrigel (Corning, 356255) -coated plates and cultured in complete astrocyte medium (#1801, ScienCell). After 30 days of differentiation, the astrocytes were ready for further experiments.
Statistical analysisGraphPad Prism software (version 10) or R Program (regression analysis, version 4.3.2) was used for all statistical analyses. Unpaired t-test, one-way or two-way analysis of variance (ANOVA) were used to determine statistical significance, followed by Tukey’s test for multiple comparisons. All quantitative data are presented as mean ± SD. Statistical significance was defined as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Comments (0)