
Remember me

We confirm that our research complies with all relevant ethical regulations.
Curation of publicly available datasetsWe retrieved human metagenomic data from six publicly available datasets1,2,3,4,11,16 through the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) using the accession numbers ERP127050, SRP339782, SRP197281, SRP116709 and SRP115355 (ERP104577 for RCC) using NCBI sra-tools (v2.11.2). These publicly available cohorts are shown in Supplementary Table 1 (Supplementary Table 3 for RCC). We included only those samples in our study that were collected at baseline, that is, before starting the therapy. We excluded any samples from patients who received combination therapy (that is, anti-CTLA4 along with anti-PD1 or IFNγ with anti-PD1) and/or had a recent history of proton pump inhibitor, H2 receptor antagonist or antibiotic usage whenever metadata for these parameters was available. We classified patients into responder and non-responder groups according to the outcomes reported in the respective studies above. Patients classified as having stable disease, partial response or complete response were considered responders, and patients with progressive disease were considered non-responders for the purposes of our analyses.
We are aware that working with public datasets may introduce biases and artificial variability into the analyses due to nonuniformity of the data structure. It is noteworthy that in our study we did not directly use the processed count or abundance matrices, which are known to contribute to batch effects. Along with the baseline filtering criteria mentioned above, we also implemented a uniform stringent quality control and downstream analysis method for all the samples at individual raw sequencing read levels, as detailed below. Thus, all the pooled sequencing samples were treated as part of one experiment, and the source of the sample (that is, study and country) was used as a covariate, similar to the original studies.
Quality control and preprocessingThe pre-processing pipeline consists of three main stages: (1) initial quality control by removing low-quality reads (quality score <Q30), reads <75 bp and Illumina adapters from the forward and reverse reads using Trimmomatic software (v0.39)33; (2) DNA contamination removal using Bowtie 2 (v2.3.5.1)34 in sensitive mode, removing host DNA from humans (GRCh38) and mice (GRCm39) and non-host phiX174 Illumina spike-in; and (3) checking the FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) results and removing any samples that had >50% read duplication from the human-associated metagenomes. FastQC results were aggregated with multiqc (v1.11) for easy manual checking.
Contig assembly, gene-calling and functional annotationQuality-trimmed host decontaminated paired-end metagenomic reads were assembled individually using metaSPAdes (v3.15.4)35 to build contigs. Single-end samples (RCC and cohorts; Supplementary Table 3) were assembled using MEGAHIT (v1.2.9)36. Contigs from individual samples were concatenated into a single file, and contigs smaller than 500 bp were discarded. To make a non-redundant contig database, 100% identical contigs were clustered, and a representative longer contig was kept using cd-hit-est from CD-HIT program (v4.8.1)37,38. Prodigal (v2.6.3)39 was used in ‘meta’ mode for the ORF and translated protein prediction from the non-redundant contig collection. The script sqm_annot.pl from SqueezeMeta software (v1.5.2)40 was used to assign KEGG (Kyoto Encyclopedia of Genes and Genomes) orthology and corresponding microbial taxa to the predicted protein sequences. The databases were downloaded, using the script make_databases.pl from SqueezeMeta. In brief, functions were assigned using Diamond (v2.0.15.153)41 blastp alignments of the proteins against the KEGG42 and NCBI non-redundant43 databases using the lowest common ancestor44 methods. This way each protein sequence from the metagenomes was assigned to a function (KEGG) and bacterial taxa, and thus a connection between function and taxonomy was established. To quantify the predicted genes in the samples across the metagenomes, we used the Salmon tool (v1.8.00)45. First, the predicted ORFs were indexed, and then reads (paired end or single end) from individual samples were mapped onto the indexed Salmon database.
Analyses of patient metagenomesMetagenomes from patients with melanoma or RCC were curated and quality controlled as described above. For gene-level relative abundance, undetected (zero value) samples were excluded, and all non-zero values were reported. In patients with RCC, the sample numbers for hexa-acylated LPS were as follows: lpxM, n = 44; lpxJ, n = 50; and total (lpxM + lpxJ), n = 51. A KEGG orthologous group present in less than five samples was considered as sporulating and/or misannotation and removed from the analysis. Relative abundance of the predicted LPS biosynthetic genes (ORFs) from the patient metagenomes was compared between ‘Responder’ and ‘Non-responder’ through a single comparison, and therefore there was no need for a multiple testing correction. Before functional analysis, we used ComBat-Seq (v3.48.0)46 to correct the batch effects from the functional profile of the cohort of patients with melanoma. ComBat-Seq was run on the read count matrix with study metadata as batch and treatment response as group (full model).
Taxonomic profiling was performed with Kraken v2.1.247 and Bracken v2.6.2 (https://doi.org/10.7717/peerj-cs.104) using a custom database built from representative genomes of the human gut microbiota that were re-annotated using the Genome Taxonomy Database v2.1. This database is available via Zenodo at https://doi.org/10.5281/zenodo.7319344 (ref. 48). After analysis with Kraken2/Bracken2, we used ConQuR (v1.2.0)49 to correct for batch effects between studies. ConQuR was run on the OTU read count matrix using a penalized fitting strategy (logistic_lasso=T, quantile_type=“lasso”, interplt=T) with study metadata as the batch variable and the response as a covariate. The resulting batch-corrected read count matrix was analysed in R (v4.3.1).
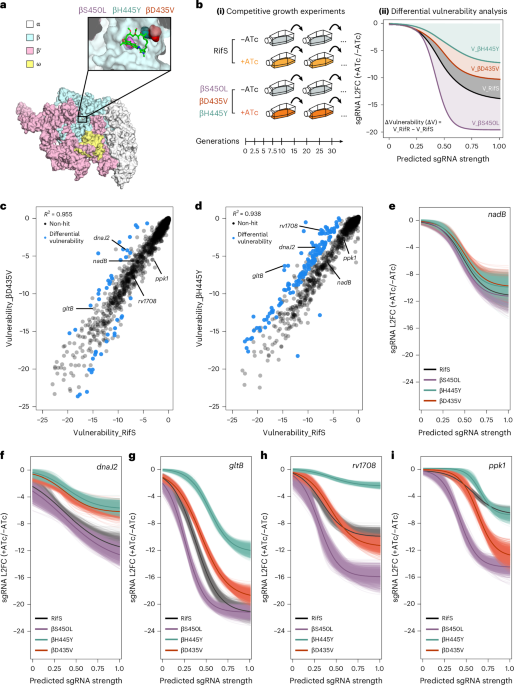
Genomic prediction of bacterial LPS structureTo predict which bacterial species of the human gut microbiota produce LPS, lpx operon genes were identified from functional pangenomes of 3,006 bacterial species using the ‘feature_search’ module of the MGBC-Toolkit v1.017. lpx genes were identified using both the InterPro and EggNOG functional schemata. Accessions for searched lpx genes are shown in Supplementary Table 4.
For each pangenome, genes that were encoded by at least 50% of the constituent genomes were considered to be encoded by that bacterial species. As most genes could be annotated using both InterPro and eggNOG, species that were identified as encoding a gene by at least one schema were included in analyses. All annotations were manually reviewed in context of the published literature and known structures for LPS. Following this review, only the InterPro database was used for lpxM annotation as eggNOG over-identified these genes in taxa which are known not to encode them.
LPS structure was then predicted using these annotation data according to the KEGG reference pathway for LPS biosynthesis. Species encoding lpxA, C, D, B and K were considered to produce lipid IVA and were thus defined as total Gram-negative LPS-encoding taxa (n = 884), while taxa that additionally encoded kdtA/waaA were considered to produce KDO2-lipid IVA (n = 827). Taxa that further encoded lpxL and either lpxM or lpxJ were considered to have the capacity to produce hexa-acylated LPS (lpxLM, n = 127; lpxLJ, n = 112), while taxa that further encoded lpxL but not lpxM or lpxJ were defined as penta-acylated LPS producers (n = 538). LPS-encoding taxa that did not encode lpxL were defined as tetra-acylated LPS producers (n = 107).
Data normalizationGene abundance of the single melanoma1, and RCC cohort was expressed in the form of gene count per million (GCPM).
The GCPM within the sample was calculated as follows:
$$}}_=\frac_/_\right)}_\left(_/_\right)}\times ^$$
where qi denotes the number of raw reads mapped to the predicted gene, li is the gene length, and ∑j(qj/lj) corresponds to the sum of mapped reads to the predicted gene normalized by gene length. Thus, GCPM accounts for gene length and sequencing depth and facilitates comparisons across samples. As the batch correction was performed on the raw read counts of the pooled cohort of patients with melanoma, we used a slightly different method to normalize and scale our data. The batch-corrected count matrix was log-transformed and added with a pseudo count of 10−6 followed by scaling to count per million (CPM) value.
The CPM within the sample was calculated as follows:
$$}}_=\frac_}_^_}\times ^$$
where Ai is the batch-corrected number of reads mapped to the predicted gene and n is the total number of predicted genes.
Clustering and functional enterotype analysisFunctional enterotyping and between-class analyses were performed according to the procedure described by Arumugam et al.50 Data generated by functional annotation with the KEGG database (KEGG orthologs) were used to calculate the JSD among samples. The PAM clustering algorithm was applied to cluster the relative abundance of functional profiles of genus or orthologous groups. The number of optimal clusters was estimated using the Calinski–Harabasz index using the clusterSim package in R according to a previously described method51. The between-class analysis was performed to identify the major drivers of the metagenome functions and support clustering the orthologous groups abundance profiles into clusters. The between-class analysis is a type of ordination with instrumental variables with the highest effect sizes that maximize the separation between classes of this variable. In this study, the instrumental variable was cluster classification using PAM clustering, obtained from KEGG orthologous groups, which contributed the maximum to the principal coordinates, identified based on their eigenvalues. The analysis was performed using the ade4 package in R.
MiceIn vivo tumour experiments were performed using 9-week-old female C57BL/6 mice purchased from Charles River. Experiments were conducted in the University of Cambridge UBS Anne McLaren facility in accordance with UK Home Office guidelines and were approved by the University of Cambridge Animal Welfare and Ethical Review Board (PPL number PP3638130). Mice were kept under a consistent 12 h light–dark cycle (7 a.m. to 7 p.m.), ambient temperature (20–24 °C) and humidity 45–65%, as per the UK Home Office guidelines. All mice were fed Scientific Diets SAFE 105 rodent diet ad libitum.
Treatment of mice with antibioticsMice were treated with PMB (500 mg l−1) (APExBio, C3090-APE) or a broad-spectrum antibiotic solution (ABX: ampicillin (1 mg ml−1) (Cayman Chemical, 14417-25g-CAY), streptomycin (3 mg ml−1) (Sigma-Aldrich, S9137-25G), and colistin (1 mg ml−1) (BioServ UK Limited, BS-9867A-5G)) in sterile drinking water. Treatment was started 2 weeks before tumour implantation, and antibiotic water was changed twice a week.
Shotgun sequencing of mouse metagenomesMouse faecal pellets were collected immediately after arrival from the supplier, termed ‘Pre’ samples. Additional faecal samples were collected after 2 weeks of treatment with broad-spectrum antibiotics, PMB or control drinking water. For broad-spectrum antibiotic-treated mice, treatment was discontinued for 1 day before sample collection to allow for some bacterial DNA to be recovered. A detailed description of the mouse metagenomic samples has been provided in Supplementary Table 5. Mouse faecal genomic DNA was extracted using the FastDNA SPIN kit for soil (MP Biomedicals, 116560200) per the manufacturer’s instructions and stored in a −80 °C freezer. Shotgun metagenomic sequencing was carried out at GENEWIZ from Azenta Life Sciences on an Illumina NovaSeq -X platform. Each sample was sequenced at a minimum depth of 20 million reads (minimum 10 million paired-end reads) with a 150-nucleotide read length. Sequences are deposited in NCBI SRA under Bioproject ID PRJNA1171992.
Taxonomic classification of mouse gut metagenomesFiltered paired-end metagenomic reads were used for taxonomic profiling of mouse gut metagenomes with Kraken v2.1.2 and Bracken v2.6.2 using the Mouse Gastrointestinal Bacteria Catalogue (MGBC) database17, as described above. To predict the LPS-producing bacterial species in the mouse metagenomes, lpx operon genes were identified using the MGBC protein database17. The abundance data were normalized by the weight of the faecal pellet used for the DNA extraction. Data were analysed and visualized in R.
Mass spectrometry of acylation of lipid A from commensal gut bacteriaMass spectrometry analyses of lipid A were performed using an Agilent 1290 Infinity II LC system coupled to an Agilent 6546 Q-TOF instrument. Lipid A-1P standard (Sigma-Aldrich catalogue number L6638-1MG) was prepared in chloroform/methanol/water (74:23:3) at 1 mg ml−1. About 5 µl of 0.5 mg ml−1 of lipid A-1P was injected onto a Waters XBridge C18 column (50 mm × 2.1 mm, 3.5 µm particle size), and the mobile phase was a linear gradient of 0–95% isopropyl alcohol with 200 mM ammonium hydroxide and 5 µM medronic acid (mobile phase B) in methanol/water (80:20) with 200 mM ammonium hydroxide and 5 µM medronic acid (mobile phase A) over 17 min (before holding at 100% mobile phase A for 3 min). The flow rate was 0.25 ml min−1, and the column was operated at 45 °C. Post-column addition of 10% DMSO in acetone at a rate of 0.25 ml min−1 was used. The published extracted ion chromatograms by Sándor et al.52 were used to characterize the lipid A molecule. Increases in acyl chain length led to later chromatographic elution.
Mouse commensal Escherichia coli (isolate A7_7.EC.CDM; NCBI assembly GCA_910574425.1) and Klebsiella pneumoniae (isolate A5_5.KP.CDM; NCBI assembly GCA_910574035.1) from our mouse culture collection were confirmed to encode lpxM using the MGBC ToolKit with ‘feature_search’ function. These isolates were inoculated in 5 ml of yeast casitone fatty acid (YCFA) broth using a single colony from streaked YCFA agar plates. The cultures were incubated anaerobically overnight at 37 °C. The 5 ml cultures were used to inoculate 1 l of YCFA liquid media. One-litre liquid cultures were incubated anaerobically overnight at 37 °C to an optical density at 600 nm of approximately 1.000. Cultures were separated into 200 ml tubes and centrifuged at 3,500 × g for 20 min. Media was discarded, and bacterial cell pellets were washed once with PBS. Dry pellets were stored at −80 °C. The culture pellet was prepared at 100 mg ml−1 in water, hydrolysed with a mild acid hydrolysis buffer (50 mM sodium acetate), and Bligh-Dyer extracted (proportion of chloroform/methanol/water, 1:2:0.8). Final resuspension was in chloroform/methanol/water (74:23:3), and 5 µl was injected onto the Waters XBridge C18 column (50 mm × 2.1 mm, 3.5 µm particle size), using the same chromatographic conditions as described above. The acyl chain lengths for the monophosphorylated and diphosphorylated forms of lipid A were annotated based on Sándor et al.52, and the proportional composition of these acyl chains was calculated for each isolate.
Tumour challenge and treatmentMC38 colon carcinoma cells were purchased from Kerafast. Cells were passaged in DMEM (Sigma-Aldrich, D5796) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich BCCC3714), 1% Glutamax (Thermo Fisher Scientific, 35050-038), 1% non-essential amino acids (Thermo Fisher Scientific, 11140-035), 1% sodium pyruvate (Thermo Fisher Scientific, 11360-039), 1% penicillin–streptomycin (Thermo Fisher Scientific, 15140-122), 0.1% of 2-mercaptoethanol (Thermo Fisher Scientific, 21985023), amphotericin B (Thermo Fisher Scientific, 15290-026) and gentamycin (Thermo Fisher Scientific, 15750-045). Mice under isoflurane anaesthesia were injected subcutaneously with 106 cells in 100 µl of sterile 1× PBS in the right flank. Tumours were measured using callipers at days 7, 10, 14, 17 and 20 after implantation. To control for incomplete tumour engraftment, mice with a tumour volume of less than 27 mm3 at day 7 after implantation were removed from the study. Mice were injected intraperitoneally (i.p.) with 200 µg anti-PD-1 (clone 29F.1A12) (BioXCell, BE0273) or IgG2a isotype control (BioXcell, BE0089) twice a week. Hexa- and penta-acylated LPS were purchased from Sigma-Aldrich (L2630-100MG and L9143-100MG). Mice were administered LPS (25 mg l−1) in sterile drinking water starting 10 days after tumour implantation. In line with our animal ethics guidelines, mice were culled if their tumour reached 1.5 cm in diameter or if the tumour became ulcerated. LPS-containing water was changed twice a week. The TLR4 inhibitor Resatorvid (TAK-242) (APExBIO, A3850) was administered i.p. on days 10, 12, 14, 17 and 19 post tumour implantation (60 μg per injection).
Sample processing of murine tumours, spleens, mesenteric lymph nodes and tumour-draining lymph nodes for immunophenotypingTumour and spleen samples were digested in Roswell Park Memorial Institute (RPMI) medium containing collagenase D and DNase I (Sigma-Aldrich, 11088882001 and 10104159001) at 37 °C for 30 min before dissociation through 70 µm cell strainers. A 40/80 Percoll gradient was used to isolate lymphocytes from tumours, and resulting cell suspensions were filtered using 40 µm cell strainers. For spleen samples, Red Blood Cell Lysis Buffer (Sigma-Aldrich, R7757-100ML) was applied at room temperature for 5 min to selectively lyse red blood cells. Mesenteric lymph nodes and tumour-draining lymph nodes were digested in complete HBSS with 2% FBS containing collagenase D at 37 °C for 30 min and dissociated through 70 µm cell strainers.
Flow cytometry of tumour-infiltrating and lymphoid tissue lymphocytesIsolated lymphocytes were re-stimulated with 1 µg ml−1 Brefeldin A (eBioscience, 00-4506-51), 50 ng ml−1 phorbol 12-myristate 13-acetate (Sigma-Aldrich, P8139-1MG) and 1 µg ml−1 ionomycin (Sigma-Aldrich, I0634-1MG) in complete RPMI with 10% FBS in a 37 °C cell culture incubator with 5% CO2 for 4.5 h. Dead cells were then stained using the LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Thermo Fisher Scientific, L34965). Cells were then stained with extracellular antibodies for 20 min on ice and permeabilized for 30 min using the BD Cytofix/Cytoperm Fixation/Permeabilization Kit (BD Biosciences, 554714) according to the manufacturer’s protocol. The intracellular antibodies listed in Supplementary Table 6 were then added and incubated for 30 min on ice. Cells were post-fixed in 1% PFA at 4 °C until analysis on a Cytek Aurora flow cytometer. Samples with cell viability below 30% were excluded from further analyses. Quantification of cell numbers was achieved using CountBright Absolute Counting Beads (Thermo Fisher Scientific, C36950), in accordance with the protocol provided by the manufacturer. Data were exported as FCS files using Cytek Aurora software (v3.3.0) and analysed using FlowJo software (v10.8.1, Tree Star). For lymphocyte flow cytometry antibody panel, see Supplementary Table 6.
Flow cytometry of myeloid cellsThe TruStain FcX Antibody (BioLegend, 101320) was used to block cells for 5 min at 4 °C. Dead cells were then stained using the LIVE/DEAD Fixable Aqua Dead Cell Stain Kit. After washing, cells were stained with the surface antibodies listed in Supplementary Table 6 in 1× PBS for 20 min on ice. Cells were fixed in 1% PFA at 4 °C until analysis on a Cytek Aurora flow cytometer. Data were exported as FCS files using Cytek Aurora software (v3.3.0) and analysed using FlowJo software (v10.8.1, Tree Star). The myeloid flow cytometry antibody panel is shown in Supplementary Table 6.
In vitro NF-kB activation in monocyte/macrophagesTHP-1 cells (THP1-Dual Reporter Cells, TLR2 KO Dual Reporter THP-1 Cells, TLR4 KO Dual Reporter THP-1 Cells and THP1-Dual MD2-CD14-TLR4 over-expressing THP-1 cells) were purchased from InvivoGen. Cells were cultured in RPMI 1640 (Gibco) with 2 mM l-glutamine (Sigma-Aldrich), 25 mM HEPES (Gibco), 10% FBS (Sigma-Aldrich), 100 µg ml−1 Normocine (InvivoGen), 100 U ml−1 penicillin–streptomycin (Gibco), 10 µg ml−1 blasticidin (InvivoGen) and 100 µg ml−1 Zeocin (InvivoGen) according to the manufacturer’s instructions. For NF-kB activation assays, THP-1 cells were resuspended in Test medium (RPMI 1640 with 2 mM l-glutamine, 25 mM HEPES, 10% FBS and 100 U ml−1 penicillin–streptomycin) and cultured at 105 cells per well for 24 h in a final volume of 200 µl of Test medium with indicated concentrations of ultrapure LPS from E. coli (hexa LPS, InvivoGen) and/or ultrapure LPS from Porphyromonas gingivalis (penta LPS, InvivoGen) with or without PMB (APExBio) in CELLSTAR 96-well plates (Greiner). About 20 µl−1 of culture supernatants was mixed with 180 µl of QUANTI-Blue Solution (InvivoGen) in 96-well enzyme-linked immunosorbent assay microplates (Greiner) according to the manufacturer’s instructions. The plates were incubated at 37 °C for 1–6 h. Optical density at 630 nm was measured on a FLUOstar Omega (BMG Labtech).
In vitro cytokine secretion in monocyte/macrophagesHuman THP-1 cells were cultured as indicated above, and supernatants were collected at 24 h. The indicated cytokine cytometric beads and paired-end-conjugated detection antibodies from the BD Cytometric Bead Array Human Inflammatory Cytokines Kit (BD Bioscience) were mixed with these supernatants according to the manufacturer’s instructions. Mouse RAW 264.7 cells (Merck, catalogue number 91062702-1VL) were cultured in high-glucose DMEM (Sigma-Aldrich) with 10% FBS (Sigma-Aldrich) and 100 U ml−1 penicillin–streptomycin (Gibco). About 105 cells per well were cultured for 24 h in a final volume of 200 µl culture medium with indicated concentrations of ultrapure LPS from E. coli (hexa LPS, InvivoGen) and/or ultrapure LPS from P. gingivalis (penta LPS, InvivoGen) with or without PMB (APExBio) in CELLSTAR 96-well plates (Greiner). Culture supernatants were mixed with the indicated cytokine beads and paired-end-conjugated detection antibodies from the Mouse Th1/Th2/Th17 CBA Kit (BD Bioscience) according to the manufacturer’s instructions. All samples were analysed on a BD LSRFortessa Cell Analyzer (BD Bioscience).
Statistical analysisStatistical analyses were performed using either Graphpad Prism software (v9.3.1) or R (v4.3.1). Non-parametric Kruskal–Wallis tests and parametric Brown–Forsythe tests were used as indicated to calculate statistical significance of the difference in sample medians and means, respectively, and post hoc Dunn P values with Bonferroni false discovery rate adjustment were calculated for the pairwise comparisons among the enterotypes. The proportions of clinical responders were compared between enterotypes using one-tailed two proportions Z-tests without continuity correction. Two-way analysis of variance (ANOVA) was used to evaluate statistical significance in experiments with tumour growth measurements. We used NMDS, a distance-based ordination technique53,54 in R (vegan::metaMDS()) with default engine = “monoMDS”. In the process of calculation of goodness of fit, a single NMDS run identifies a local minimum; therefore, we iterated the runs 1,000 times, or the best solution achieved to reach the global minimum (convergence) using the function Procrustes in R (vegan:: procrustes()). The final stress score was used to assess the optimal model and goodness of fit as suggested previously55,56. In addition to the stress score, we also performed a two-tailed t-test on the NMDS coordinates as shown previously57 to highlight the difference between the ‘Responder’ and ‘Non-responder’ groups. P values of less than 0.05 were considered statistically significant. Statistical tests used are specified in the figure legends. Data distribution was assumed to be normal/parametric or non-parametric based on the data type, but this was not formally tested. P values correlate with symbols as follows: NS, not significant; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. No statistical methods were used to pre-determine sample sizes, but where relevant, sample sizes were determined based on variability observed in published experiments of a similar kind2. In some experiments prior experience of sample size requirement was used to design experimental group sizes. For experiments where technical limitations prevented adequate statistical power to be obtained from single experiments, results from multiple experiments were pooled to provide sufficient statistical power. Data reported are in most cases non-subjective and did not require randomization or blinding at measurement. Where possible, data collection and/or the organization of the experimental conditions were randomized. Investigators were not formally blinded. However, in randomized experiments it was difficult for investigators and technicians to readily determine treatment groups from animal IDs at the bench.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Comments (0)